Снип технологическое оборудование и технологические трубопроводы: СНиП 3.05.05-84 Технологическое оборудование и технологические трубопроводы
ТЕХНОЛОГИЧЕСКОЕ ОБОРУДОВАНИЕ И ТЕХНОЛОГИЧЕСКИЕ ТРУБОПРОВОДЫ
ГОСУДАРСТВЕННЫЙ КОМИТЕТСССР
ПО ДЕЛАМ СТРОИТЕЛЬСТВА
РАЗРАБОТАНЫВНИИмонтажспецстроем Минмонтажспецстроя СССР (инж. В. Я. Эйдельман, д-р техн. наук В. В. Поповский — руководители темы; кандидаты техн. наук В. И. Оботуров, Ю. В. Попов, Р. И. Тавастшерна), Гипронефтеспецмонтажом Минмонтажспецстроя СССР (канд. техн. наук И. С. Гольденберг) и Гипрохиммонтажом Минмонтажспецстроя СССР (инженеры И. П. Петрухин, М. Л. Эльяш).
ВНЕСЕНЫ Минмонтажспецстроем СССР.
ПОДГОТОВЛЕНЫ К УТВЕРЖДЕНИЮ Отделом технического нормирования и стандартизации Госстроя СССР (инж. Б. А. Соколов) .
С введением в действие СНиП 3.05.05-84 „Технологическое оборудование и технологические трубопроводы» утрачивает силу СНиП III. 31.78*„Технологическое оборудование. Основные положения”.
| Государственныйкомитет по делам строительства (Госстрой СССР) | Строительные нормы и правила | СНиП 3. 05.05-84 05.05-84 |
| Технологическое оборудование и технологические трубопроводы | Взамен СНиП III-31-78* |
Настоящие правила распространяются на производство и приемку работ по монтажу технологического оборудования и технологических трубопроводов (в дальнейшем — „оборудование» и „трубопроводы»), предназначенных для получения, переработки и транспортирования исходных, промежуточных и конечных продуктов при абсолютном давлении от 0,001 МПа (0,01 кгс/см2) до 100 МПа вкл. (1000 кгс/см2), а также трубопроводов для подачи теплоносителей, смазки и других веществ, необходимых для работы оборудования.
Правила должны соблюдаться всеми организациями и предприятиями, участвующими в проектировании и строительстве новых, расширении, реконструкции и техническом перевооружении действующих предприятий.
Работы по монтажу оборудования и трубопроводов, подконтрольных Госгортсхнадзору СССР, в том числе сворка и контроль качества сварных соединений, должны производиться согласно правилам и нормам Госгортехнадзора СССР.
Общие положения
Подготовка к производству монтажных работ
Производство монтажных работ
Сварные и другие неразъемные соединения трубопроводов
Индивидуальные испытания смонтированного оборудования и трубопроводов
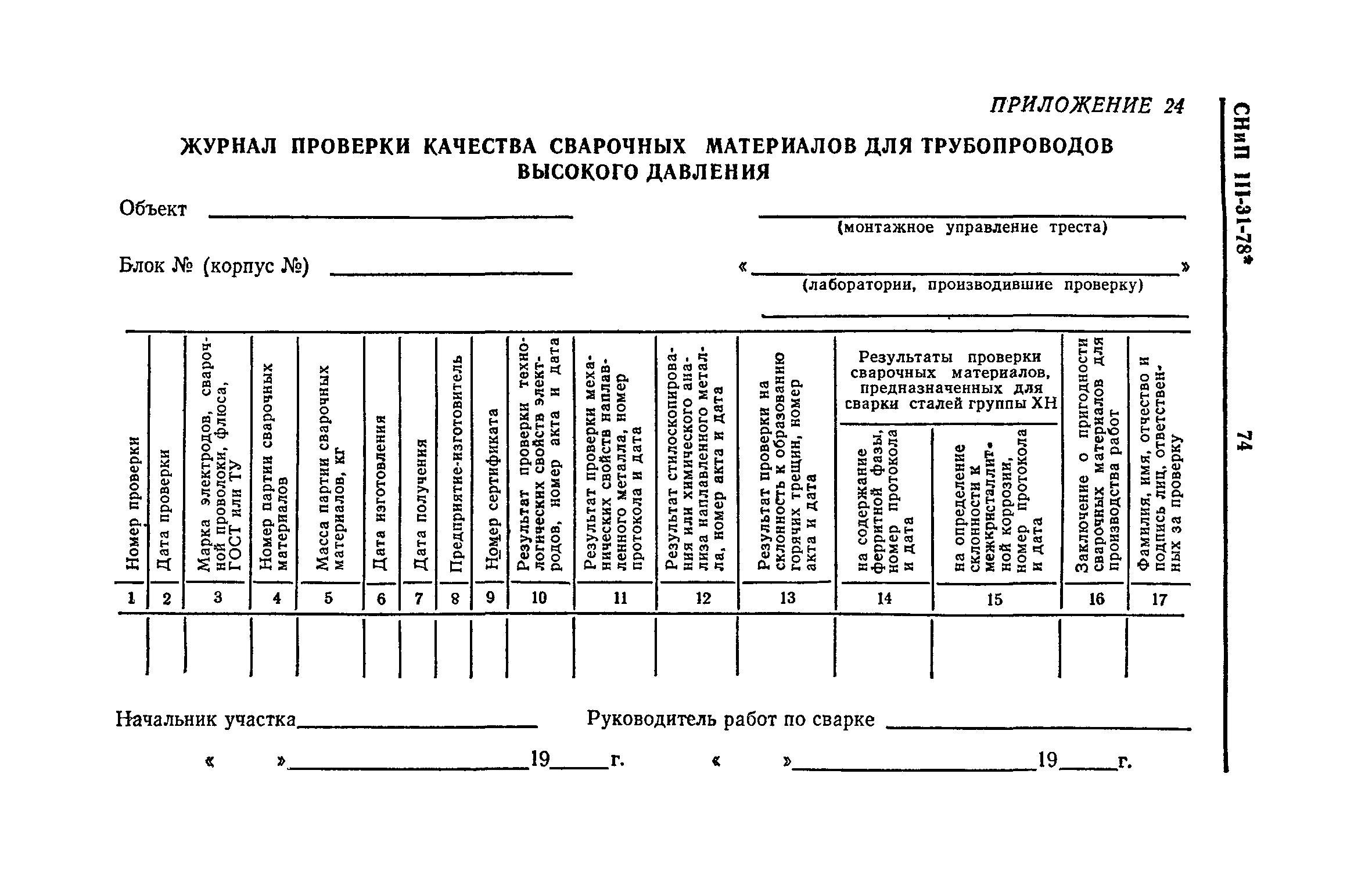
Приложение 1
Приложение 2
Приложение 3
Приложение 4
Технологическое оборудование и технологические трубопроводы- строительные нормы и правила- СНиП 3-05-05-84 (утв- постановлением Госстроя СССР от 07-05-84 72). Актуально в 2019 году
Наименование документа:
«ТЕХНОЛОГИЧЕСКОЕ ОБОРУДОВАНИЕ И ТЕХНОЛОГИЧЕСКИЕ ТРУБОПРОВОДЫ. СТРОИТЕЛЬНЫЕ НОРМЫ И ПРАВИЛА. СНиП 3.05.05-84» (утв. Постановлением Госстроя СССР от 07.05.84 N 72)
Вид документа
- нормы
- порядок
- постановление
- правила
Принявший орган
- ГОССТРОЙ СССР
Номер документа
СНИП 3.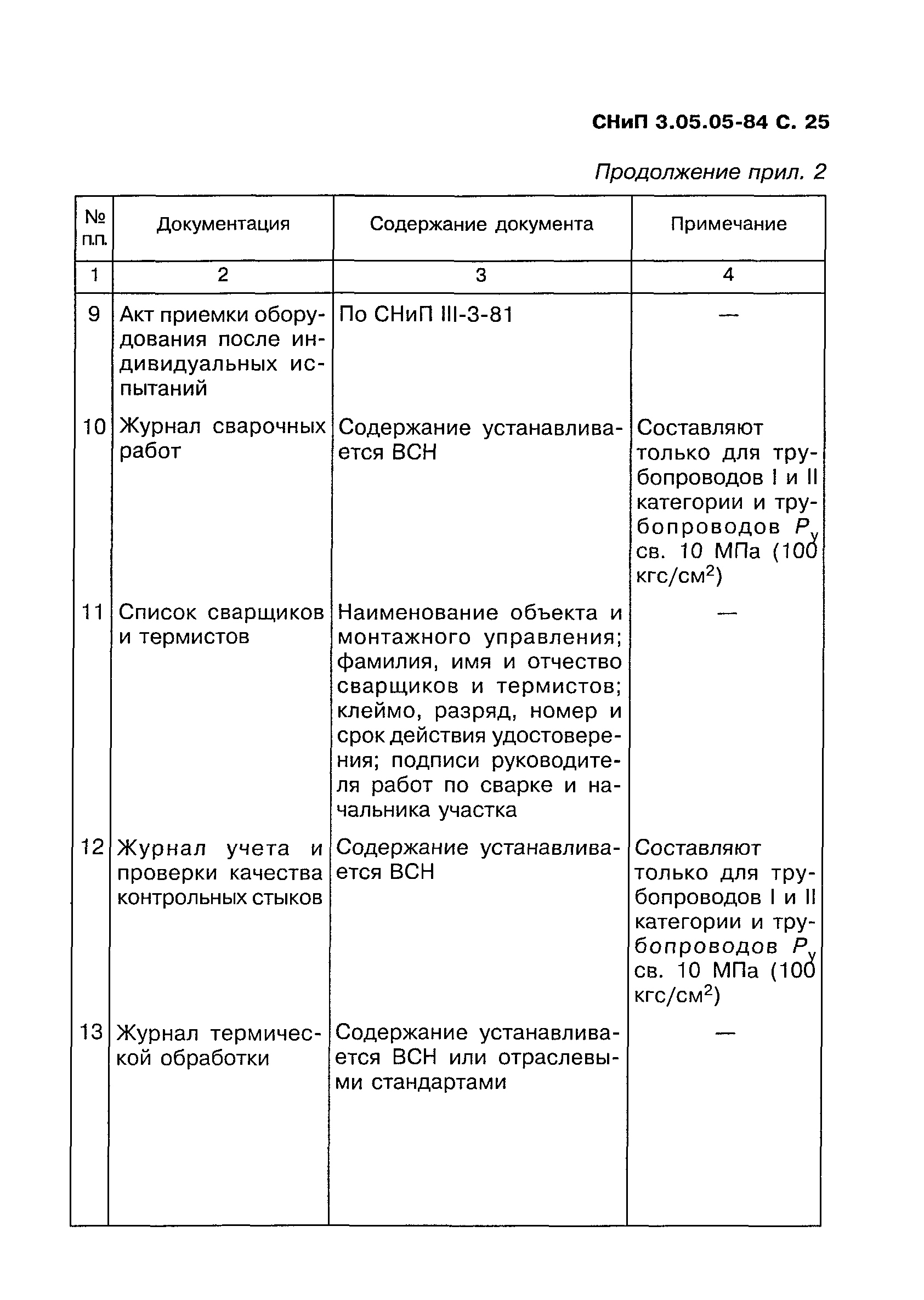 05.05-84
05.05-84
Дата принятия
Дата редакции
1984-05-07
Дата регистрации в Минюсте
Статус
Действует
Публикация
- ГП ЦПП, Москва, 1996
УТВЕРЖДЕНЫ
Постановлением Госстроя СССР
от 7 мая 1984 г.
N 72
Дата введения — 1 января 1985 г.
Настоящие правила распространяются на производство и приемку работ по монтажу технологического оборудования и технологических трубопроводов (в дальнейшем — «оборудование» и «трубопроводы»), предназначенных для получения, переработки и транспортирования исходных, промежуточных и конечных продуктов при абсолютном давлении от 0,001 МПа (0,01 кгс/кв.
Правила должны соблюдаться всеми организациями и предприятиями, участвующими в проектировании и строительстве новых, расширении, реконструкции и техническом перевооружении действующих предприятий.
Работы по монтажу оборудования и трубопроводов, подконтрольных Госгортехнадзору СССР, в том числе сварка и контроль качества сварных соединений, должны производиться согласно правилам и нормам Госгортехнадзора СССР.
Приложение 1
Обязательное
Error 404 — Законодательство, нормативные акты, образцы документов
1. «Орал
Орал қалалық мәслихатының 2014 жылғы 25 қарашадағы № 30-5 «Орал қаласында аз қамтамасыз етілген отбасыларға (азаматтарға) тұрғын үй көмегін көрсетудің мөлшерін және тәртібін айқындау туралы қағидасын бекіту туралы» шешіміне өзгерістер енгізу туралы Батыс Қазақстан облысы Орал қалалық мәслихатының 2015 жылғы 3 желтоқсандағы № 39-3 шешімі Қазақстан Республикасының 2001 жылғы 23 қаңтардағы «Қазақстан Республикасындағы жергілікті мемлекеттік басқару және өзін-өзі басқару туралы» және 1997 жылғы 16 сәуірдегі «Тұрғын үй қатынастары туралы» Заңдарына сәйк Далее. ..
..2. «
«Қазақстан Республикасы ұлттық қауіпсіздік комитеті органдарының әскери, арнаулы оқу орындарында іске асырылатын жоғары және жоғары оқу орнынан кейінгі білім беру мамандықтары бойынша үлгілік оқу жоспарларын бекіту туралы» Қазақстан Республикасы Ұлттық қауіпсіздік комитеті Төрағасының 2016 жылғы 13 қаңтардағы № 9/ҚБП бұйрығына өзгерістер енгізу туралы» Қазақстан Республикасы Ұлттық қауіпсіздік комитеті Төрағасының 2016 жылғы 10 қазандағы № 67/ҚБП бұйрығы. Қызмет бабында пайдалануға арналған және Деректер базасына енгізілмейді Далее…3. Утверждены Правила согласования размещения предприятий и других сооружений, а также условий производства строительных и других работ на водных объектах, водоохранных зонах и полосах (аннотация к документу от 01.09.2016)
Утверждены Правила согласования размещения предприятий и других сооружений, а также условий производства строительных и других работ на водных объектах, водоохранных зонах и полосахАннотация к документу: Приказ Заместителя Премьер-Министра Республики Казахстан — Министра сельского хозяйства Республики Казахстан от 1 сентября 2016 года № 380 «Об утверждении Правил согласования размещения предприятий и других сооружений, а также условий производства строительных и других работ на водных объектах, водоохранных зонах и полосах»В соответствии с подпунктом 7-5) пункта 1 статьи 37 Водного кодекса Республики Казахстан от 9 июля 2003 год Далее.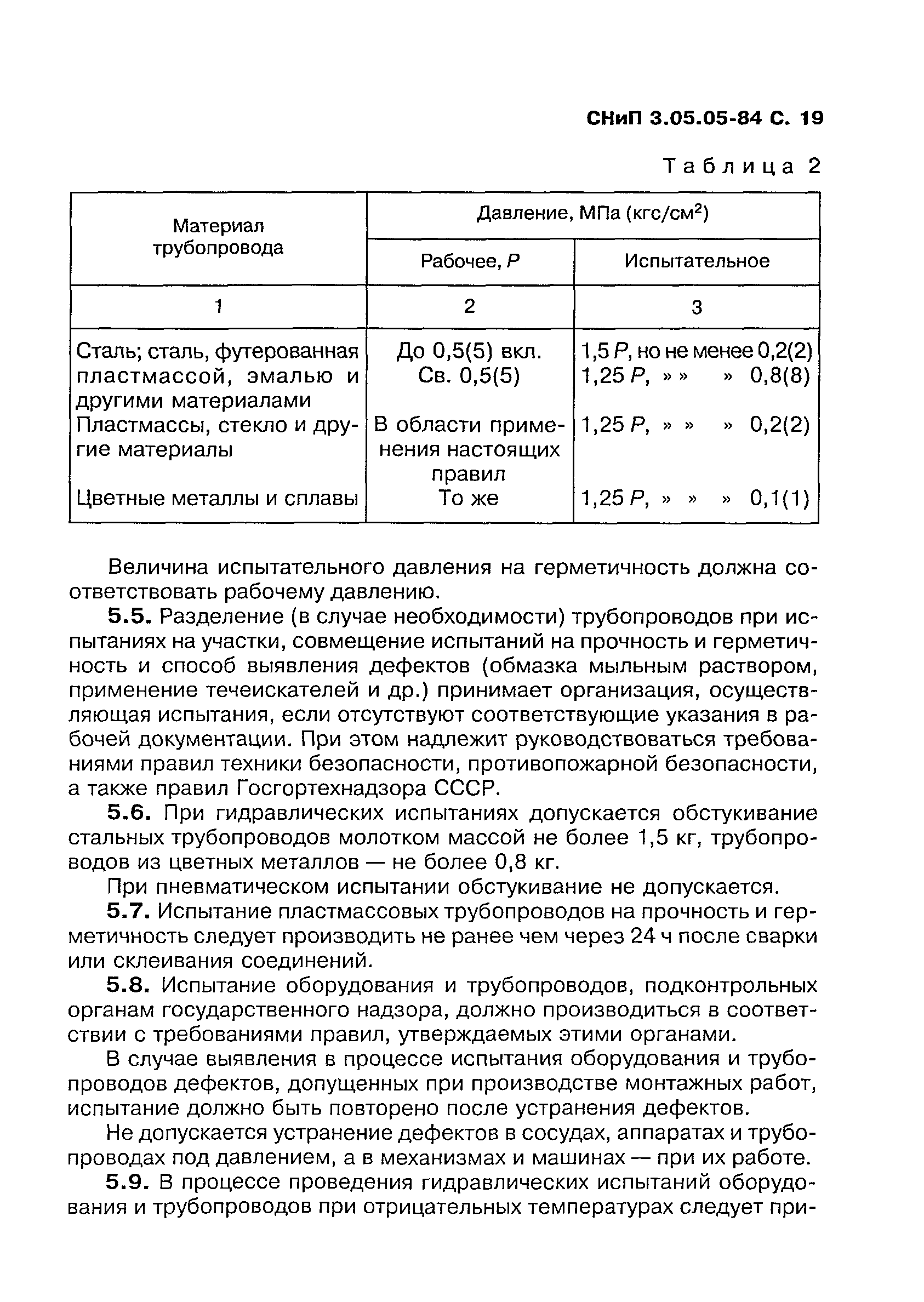 ..
..4. Утверждены Правила регулирования цен на услуги, производимые и реализуемые субъектами государственной монополии в области связи (аннотация к документу от 24.10.2016)
Утверждены Правила регулирования цен на услуги, производимые и реализуемые субъектами государственной монополии в области связиАннотация к документу: Приказ Министра информации и коммуникаций Республики Казахстан от 24 октября 2016 года № 221 «Об утверждении Правил регулирования цен на услуги, производимые и реализуемые субъектами государственной монополии в области связи»В соответствии с подпунктом 1) пункта 2 статьи 20 Закона Республики Казахстан от 5 июля 2004 года «О связи» утверждены Далее…5. Утверждены Правила формирования перечня энергопроизводящих организаций, использующих возобновляемые источники энергии (аннотация к документу от 09.11.2016)
Утверждены Правила формирования перечня энергопроизводящих организаций, использующих возобновляемые источники энергииАннотация к документу: Приказ Министра энергетики Республики Казахстан от 9 ноября 2016 года № 482 «Об утверждении Правил формирования перечня энергопроизводящих организаций, использующих возобновляемые источники энергии»В соответствии с подпунктом 10-3) статьи 6 Закона Республики Казахстан от 4 июля 2009 года «О поддержке использования возобновляемых источников энергии» утверждены Далее.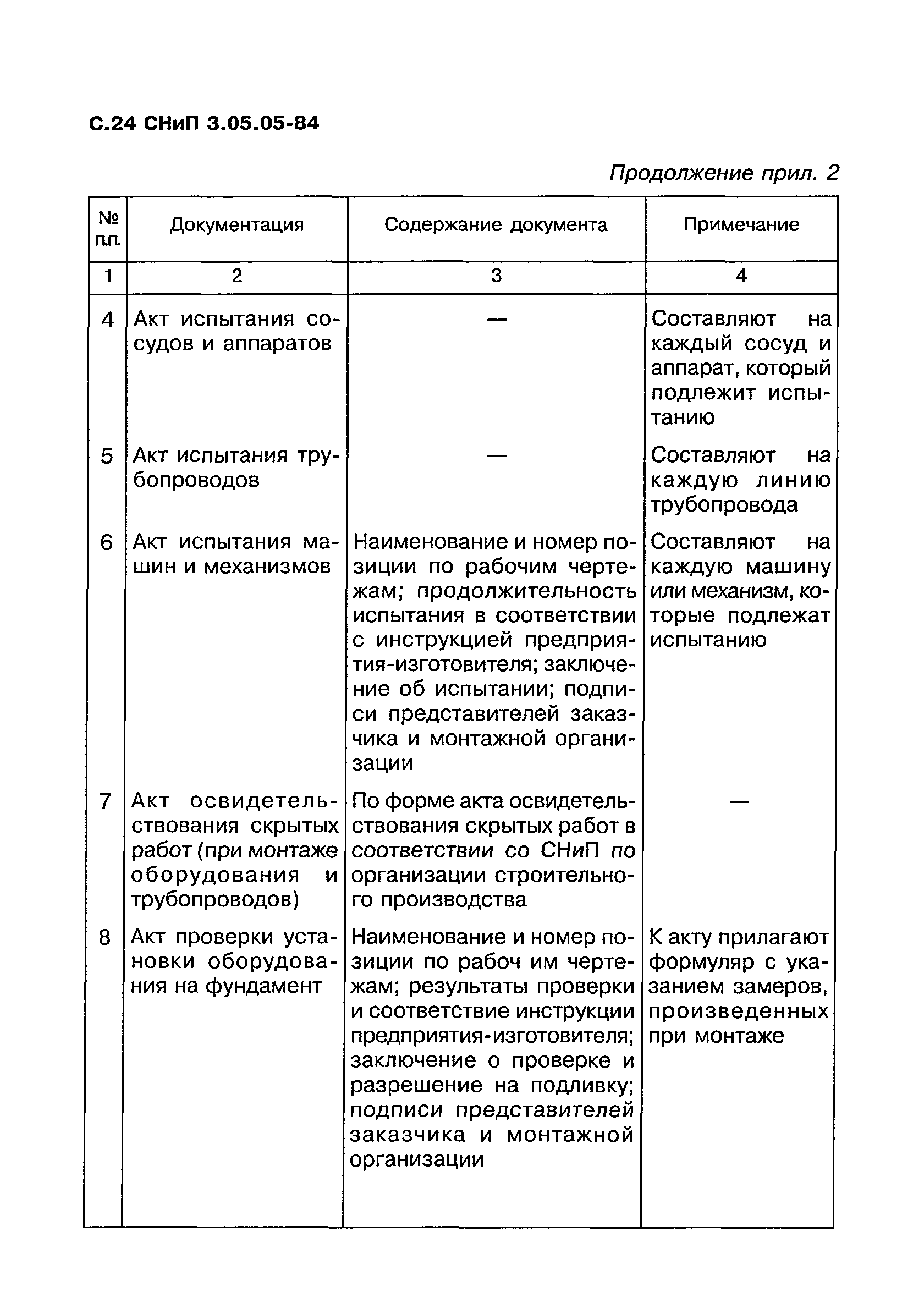 ..
..6. Изменения внесены в ряд приказов Министра энергетики Республики Казахстан (аннотация к документу от 31.05.2016)
Изменения внесены в ряд приказов Министра энергетики Республики КазахстанАннотация к документу: Приказ Министра энергетики Республики Казахстан от 31 мая 2016 года № 228 «О внесении изменений в некоторые приказы Министра энергетики Республики Казахстан»В частности, изменения внесены в приказ Министра энергетики Республики Казахстан «Об утверждении Правил пользования тепловой энергией», изменения затронули понятия и определения используемые в правилах. Также, изменения внесены в ряд пунктов правил, а именно: Далее…7. Заканчивается срок приема заявлений по легализации имущества
Заканчивается срок приема заявлений по легализации имущества Вниманию всех заинтересованных лиц!Напоминаем, что 31 декабря 2016 года заканчивается легализация имущества, которая проводилась с 1 сентября 2014 года в соответствии с Законом РК от 30 июня 2014 года № 213-V «Об амнистии граждан Республики Казахстан, оралманов и лиц, имеющих вид на жительство в Республике Казахстан, в связи с легализацией ими имущества». При этом, срок подачи документов для легализации недвижимого имущества, находящегося на территории Республики Казахстан, заканчился 30 ноября 2016 года, а для иного имущества срок подачи документов завершается за 5 рабочих дней до конца 2016 года, то есть не позднее 23 декабря 2016 года. Далее…
При этом, срок подачи документов для легализации недвижимого имущества, находящегося на территории Республики Казахстан, заканчился 30 ноября 2016 года, а для иного имущества срок подачи документов завершается за 5 рабочих дней до конца 2016 года, то есть не позднее 23 декабря 2016 года. Далее…8. 31 декабря истекает срок уплаты налога на транспорт физическими лицами
31 декабря истекает срок уплаты налога на транспорт физическими лицами Вниманию физических лиц, имеющих на праве собственности транспортные средства!Срок уплаты налога на транспортные средства истекает 31 декабря 2016 года.Обратите внимание, что с 1 января 2016 года уплата налога физическими лицами производится по месту жительства.В случае осуществления регистрационных действий по передаче права собственности на транспортное средство, сумма налога, подлежащая уплате за фактический период владения таким объектом лицом, передающим эти права, должна быть внесена в бюджет до совершения указанных действий.Уплата налога на транспортные средства физическим лицом, являю Далее. ..
..9. О дифференциации доходов и расходов населения в Республике Казахстан за 3 квартал 2016 года
О дифференциации доходов и расходов населения в Республике Казахстан за 3 квартал 2016 года По результатам выброчного обследования домашних хозяйств доля населения, имеющего доходы ниже величины прожиточного минимума (уровень бедности), в Республике Казахстан в 3 квартале 2016 года составила 2,5%, по сравнению с соответствующим периодом предыдущего года оставшись на том же уровне. Вместе с тем, по-прежнему, сохраняется разрыв между уровнем бедности среди городского и сельского населения. Наибольшее значение уровня бедности в 3 квартале 2016 года зарегистрировано в Южно-Казахстанской (5,0%), Атырауской и Жамбылско Далее…10. Сагинтаев поручил акимам «удержать» инфляцию
Сагинтаев поручил акимам «удержать» инфляцию Премьер-министр РК Бакытжан Сагинтаев поручил акимам регионов работать по «удержанию» инфляции в коридоре 6-8%, передает корреспондент Zakon.kz.«В прошлый раз мы говорил о том, что необходимо оставаться в коридоре 6-8% по инфляции. 11 месяц мы грубо так провалили и вот я еще раз обращаюсь к акимам регионов, чтобы в декабре 2016 года мы удержали инфляцию с тем, чтобы остаться в коридоре 6-8%. Работу будем продолжать. На следующей неделе еще поговорим по итогам», — сказал он на заседании Правительства РК.В то же время Глава Кабмина отметил, что тенденция по росту экономики в Казахстане по итогам 11 месяцев положительная.«Мы видим, что хорошие показатели имеем, тенденция положительная. И, если мы по итогам полугодия говорили о том, что было бы хорошо, чтобы мы год Далее…
11 месяц мы грубо так провалили и вот я еще раз обращаюсь к акимам регионов, чтобы в декабре 2016 года мы удержали инфляцию с тем, чтобы остаться в коридоре 6-8%. Работу будем продолжать. На следующей неделе еще поговорим по итогам», — сказал он на заседании Правительства РК.В то же время Глава Кабмина отметил, что тенденция по росту экономики в Казахстане по итогам 11 месяцев положительная.«Мы видим, что хорошие показатели имеем, тенденция положительная. И, если мы по итогам полугодия говорили о том, что было бы хорошо, чтобы мы год Далее…11. Обзор пользователей интернет-услуг ЕНПФ за декабрь 2016 года
Обзор пользователей интернет-услуг ЕНПФ за декабрь 2016 года Количество вкладчиков, выбравших метод веб-информирования Единого накопительного пенсионного фонда, на декабрь 2016 года составляет 2,77 миллиона человек. Доля пользователей онлайн услуг ЕНПФ за год выросла с 13% до 29%.Всего за год число абонентов фиксированного интернета в РК выросло на 201 тысячу, до 2,27 миллиона. Из них 55 тысяч количество новых абонентов сельской местности, всего — 436 тысяч.За 5 лет количество интернет-абонентов в РК выросло почти вдвое — на 93%. При этом показатели села подскочили почти втрое (на 179%). Далее…
Из них 55 тысяч количество новых абонентов сельской местности, всего — 436 тысяч.За 5 лет количество интернет-абонентов в РК выросло почти вдвое — на 93%. При этом показатели села подскочили почти втрое (на 179%). Далее…12. Ликвидация организации как основание прекращения производства по гражданскому делу (Тимур Данабаев, практикующий юрист)
Ликвидация организации как основание прекращения производства по гражданскому делу Тимур ДанабаевПрактикующий юрист Подпунктом 8) статьи 277 Гражданского процессуального кодекса Республики Казахстан (далее — ГПК РК) предусмотрено, что суд прекращает производство по делу если организация, выступающая стороной по делу, ликвидирована с прекращением ее деятельности и отсутствием правопреемников. Указанные ниже вопросы свидетельствуют о наличии определенных сложностей с толкованием и практическим применением в судебной практике указанной нормы права, а также о существовании различных (нередко противоречивых) подходов к ее применению. Рассмотрим эти Далее. ..
..13. Розничная торговля за ноябрь 2016 года
Розничная торговля за ноябрь 2016 года Средний чек на городского жителя в ноябре 2016 составил 66,2 тысячи тенге — на 7,5% больше, чем годом ранее. Объем ритейла за год вырос на 9,6%, и достиг 669,1 млрд тг.В ноябре объем официальной розничной торговли составил 669,1 млрд тг — на 0,3% (+2,1 млрд тг) больше, чем в октябре, и на 9,6% (+58,7 млрд тг) больше, чем годом ранее.Примечательно, что положительную динамику обеспечили регионы, в то время как обе столицы, концентрирующие 35,5% всего ритейла по РК, в минусе по отношению к октябрю 2016.Наибольший месячный прирост отмечен в Павлодарской области (почти на треть, до 36,5 млрд тг) и Жамбылской области (+17,2%, до 20 млрд тг). Далее…14. Утвержден Генеральный план города Атырау (аннотация к документу от 29.11.2016)
Утвержден Генеральный план города Атырау Аннотация к документу: Постановление Правительства Республики Казахстан от 29 ноября 2016 года № 749 «О Генеральном плане города Атырау Атырауской области (включая основные положения)» (не введено в действие)В соответствии со статьей 19 Закона Республики Казахстан от 16 июля 2001 года «Об архитектурной, градостроительной и строительной деятельности в Республике Казахстан» и в целях обеспечения комплексного развития города Атырау Атырауской области Правительство Республики Казахстан утвержден Далее. ..
..15. Реализация кадровой политики в Национальном бюро по противодействию коррупции (аннотация к документу от 21.10.2016)
Реализация кадровой политики в Национальном бюро по противодействию коррупцииАннотация к документу: Приказ Председателя Агентства Республики Казахстан по делам государственной службы и противодействию коррупции от 21 октября 2016 года № 18 «О некоторых вопросах реализации кадровой политики в Национальном бюро по противодействию коррупции (Антикоррупционной службе) Агентства Республики Казахстан по делам государственной службы и противодействию коррупции»В соответствии с подпунктом 9) статьи 5-1, Далее…16. Особенности исполнения налогового обязательства при ликвидации и прекращении деятельности (ДГД по Восточно-Казахстанской области, 15 ноября 2016 г.)
Особенности исполнения налогового обязательства при ликвидации и прекращении деятельности Законом Республики Казахстан от 29 декабря 2014 года № 269-V «О внесении изменений и дополнений в некоторые законодательные акты Республики Казахстан по вопросам кардинального улучшения условий для предпринимательской деятельности в Республике Казахстан» внесены существенные изменения в части ликвидации предприятий и ИП, а именно, предоставлена возможность закрытия по результатам аудиторской проверки. В Кодекс Республики Казахстан «О налогах и других обязательных платежах в бюджет» (далее- Налоговый кодекс) введена новая статья 37-2 «Ос Далее…
В Кодекс Республики Казахстан «О налогах и других обязательных платежах в бюджет» (далее- Налоговый кодекс) введена новая статья 37-2 «Ос Далее…17. Вернуть в административное законодательство (Ержан Карабаев, председатель апелляционной судебной коллеги по уголовным делам Мангистауского областного суда)
Вернуть в административное законодательство Ержан Карабаев, председатель апелляционной судебной коллеги по уголовным делам Мангистауского областного суда В судебной практике казахстанских судов возникают проблемные вопросы при рассмотрении уголовных дел по уголовным проступкам и при назначении наказаний за их совершение. Далее…18. Повышая доверие к правосудию (Малик Жаркынбеков, судья Актюбинского областного суда)
Повышая доверие к правосудию Малик Жаркынбеков, судья Актюбинского областного суда VII внеочередной Съезд судей Республики Казахстан определил основные направления совершенствования деятельности судов по эффективной защите прав, свобод, достоинства и собственности граждан государства. В целом работа Съезда была нацелена на становление прочной, современной, демократической судебной системы как одной из главных составляющих развития страны, развития нашего государства в среднесрочной и дальней перспективе. Далее…
В целом работа Съезда была нацелена на становление прочной, современной, демократической судебной системы как одной из главных составляющих развития страны, развития нашего государства в среднесрочной и дальней перспективе. Далее…19. К эффективной реализации реформ (М. Рысбеков, председатель СМЭС Павлодарской области)
К эффективной реализации реформ М. Рысбеков, председатель СМЭС Павлодарской области К 25-й годовщине Независимости мы подходим с новой казахстанской мечтой, которая тождественна главной цели реализуемой нами «Стратегии-2050». К середине ХХІ века мы планируем добиться вхождения Казахстана в число 30 самых развитых государств мира.Лидер нации, выступая на XVI Съезде партии «Нур Отан Далее…20. Снизить размеры взысканий (Ермек Махметов, судья САС г. Актобе)
Снизить размеры взысканий Ермек Махметов, судья САС г. Актобе Долгое время, начиная с момента обретения Казахстаном независимости, административному законодательству, регулирующему административно-деликтные правоотношения, не уделялось должного внимания, оно являлось наследием советского времени, сохранив в себе карательно-репрессивный характер. На VI Съезде судей Главой государства были обозначены пять приоритетных задач, направленных на модернизацию судебной системы, в числе которых дальнейшее совершенствование законодательства, внедрение альтернативных способов разреш Далее…
На VI Съезде судей Главой государства были обозначены пять приоритетных задач, направленных на модернизацию судебной системы, в числе которых дальнейшее совершенствование законодательства, внедрение альтернативных способов разреш Далее…Технологическое оборудование и технологические трубопроводы НПО «Стройполимер»
Техническая поддержка и проектирование
Любой производственный процесс невозможен без специальных коммуникаций. Будь то химическое или пищевое производство, оно в обязательном порядке требует особого подхода к доставке жидкостей и газов внутри самой производственной техники и оборудования или до него.
Трубопроводы технологические
НПО «Стройполимер» предлагает трубопроводы из полипропилена повышенной химической стойкости, предназначенные для работы с агрессивными химическими веществами в химическом, фармацевтическом, пищевом и других видах производства для доставки веществ с температурой, не превышающей 95ºС.
Соответствие ГОСТ и СНиП технологических трубопроводов «Стройполимер»
Испытание технологических трубопроводов производится в соответствии с тем же ГОСТом.
При проектировании и производстве учитываются требования внутренних технических документов, а также СНиП 3.05.05-84 (Технологическое оборудование и технологические трубопроводы), ПБ «Технологические трубопроводы» № 03-585-03 и других нормативных актов, регламентирующих требования и нормы при производстве полимерных трубопроводов бытового и промышленного назначения.
Монтаж технологического оборудования и трубопроводов
НПО «Стройполимер» предлагает широкий ассортимент комплектующих, полностью совместимых с производимыми трубопроводами:
- муфты,
- угольники,
- тройники,
- крестовины,
- шаровые краны,
- вентили,
- фланцевые соединения,
- компенсационные петли,
- и много другое
Соединение труб и фитингов может производиться как путем термической сварки, так и посредством фланцевых соединений.
С нашим оборудованием любая ревизия технологических трубопроводов будет проходить успешно!
3.05.05-98-сон 01.01.1998. Технологическое оборудование и технологические трубопроводы
Ҳаммаси Кейинги таҳрирга ҳавола Олдинги таҳрирга ҳавола АПК бўйича индекслаш ҚСУК бўйича индекслаш ТТР бўйича индекслаш ҚМҚ бўйича индекслаш Ўзгартиришлар манбаси Расмий нашр манбасиТехнологические трубопроводы | КарКом — строительно-инженерная компания в Магнитогорске
Монтаж технологического оборудования и трубопроводов: особенности и требования
Трубопровод состоит из таких элементов:
— плотно соединенные трубы;
— запорно-регулирующая аппаратура;
— контрольно-измерительные приборы;
— опоры;
— подвески;
— прокладки;
— детали для крепежа;
— детали и материалы для тепловой изоляции;
— другие деталей трубопроводов.
Монтаж технологического оборудования и трубопроводов может проводиться только квалифицированными специалистами, обладающие всеми необходимыми знаниями в области монтажа и технической эксплуатации промышленного оборудования.
В частности, они должны:
— быть знакомы с устройством и условиями работы трубопроводов;
— соблюдать требования к применению различных материалов, используемых при монтаже;
— выполнять правила и специальные технологические нормы.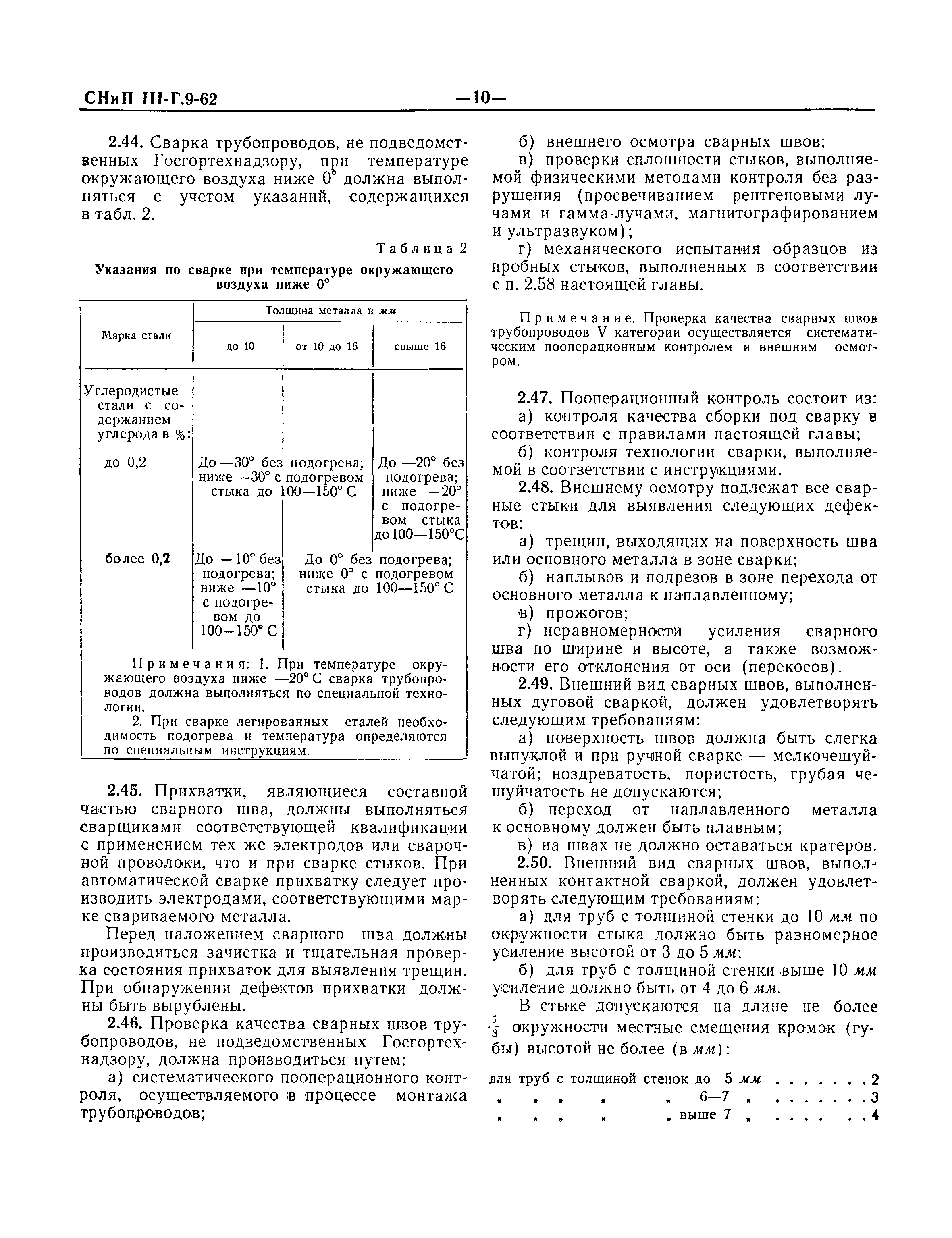
Монтаж трубопроводов и оборудования проводится на основе СНиП 3.05.05-84 «Технологическое оборудование и технологические трубопроводы».
1. Подготовка к производству монтажных работ.
- Комплектация объекта оборудованием и материалами в сроки, учитывающие последовательности монтажа, а также производства сопутствующих специальных строительных и пусконаладочных работ.
- Разработка проекта производства работ по монтажу оборудования и трубопроводов.
- Подготовка площадок для хранения оборудования и материалов, укрупнительной сборки оборудования, трубопроводов и конструкций, сборки блоков (технологических и коммуникаций).
- Подготовка грузоподъемных, транспортных средств, устройств для монтажа и индивидуального испытания оборудования и трубопроводов.
- Подготовка производственных и санитарно-бытовых зданий и сооружений.
- Выполнение необходимых мероприятий по охране труда, противопожарной безопасности и охране окружающей среды.

- Изготовление сборочных единиц трубопроводов.
- Сборка технологических блоков и блоков коммуникаций.

2. Производство монтажных работ.
- Монтаж и обвязка технологического оборудования.
- Монтаж технологических трубопроводов, а также трубопроводов для подачи воды, пара, смазки, воздуха и других веществ, необходимых для работы оборудования.
3. Соединение трубопроводов.
- Сварка стыков стальных трубопроводов, монтаж неразъемных соединений стальных, из цветных металлов и сплавов, сварка и склеивание пластмассовых трубопроводов.
- Контроль качества соединений.
4. Индивидуальные испытания смонтированного оборудования и трубопроводов.
- Гидравлические, пневматические испытания сосудов, аппаратов и трубопроводов на прочность и герметичность.
- Испытания машин, механизмов и агрегатов на холостом ходу.
5. Участие в комплексном опробовании оборудования на эксплуатационных режимах.
6. Оформление производственной документации по монтажу технологического оборудования и технологических трубопроводов.
Сравнение семи конвейеров и двух технологий секвенирования
Abstract
Секвенирование следующего поколения (NGS) произвело революцию в исследованиях растений и животных во многих отношениях, включая новые методы высокопроизводительного генотипирования. Было продемонстрировано, что генотипирование путем секвенирования (GBS) является надежным и экономически эффективным методом генотипирования, способным давать от тысяч до миллионов SNP для широкого круга видов. Несомненно, самым большим препятствием для его более широкого использования является проблема анализа данных.Здесь мы описываем всестороннее сравнение семи конвейеров биоинформатики GBS, разработанных для обработки необработанных данных о последовательности GBS в генотипы SNP. Мы сравнили пять конвейеров, требующих эталонного генома (TASSEL-GBS v1 и v2, Stacks, IGST и Fast-GBS), и два конвейера de novo , для которых эталонный геном не требуется (UNEAK и Stacks). Используя данные секвенирования Illumina из набора из 24 повторно секвенированных линий сои, мы выполнили вызов SNP с этими конвейерами и сравнили вызовы SNP GBS с данными повторного секвенирования, чтобы оценить их точность.Количество SNP, названных без эталонного генома, было ниже (от 13 000 до 24 000), чем с эталонным геномом (от 25 000 до 54 000 SNP), в то время как точность была высокой (92,3–98,7%) для всех конвейеров, кроме одного (TASSEL-GBSv1, 76,1%). . Среди конвейеров, предлагающих высокую точность (> 95%), Fast-GBS вызвал наибольшее количество полиморфизмов (около 35 000 SNP + Indels) и дал самую высокую точность (98,7%). Используя данные последовательности Ion Torrent для тех же 24 строк, мы сравнили производительность Fast-GBS с производительностью TASSEL-GBSv2.Он снова вызвал больше полиморфизмов (25,8 тыс. против 22,9 тыс.), и они оказались более точными (95,2 против 91,1%). Как правило, каталоги SNP, вызываемые из одних и тех же данных секвенирования с использованием разных конвейеров, приводили к сильному перекрытию каталогов SNP (перекрытие 79–92%).
Используя данные секвенирования Illumina из набора из 24 повторно секвенированных линий сои, мы выполнили вызов SNP с этими конвейерами и сравнили вызовы SNP GBS с данными повторного секвенирования, чтобы оценить их точность.Количество SNP, названных без эталонного генома, было ниже (от 13 000 до 24 000), чем с эталонным геномом (от 25 000 до 54 000 SNP), в то время как точность была высокой (92,3–98,7%) для всех конвейеров, кроме одного (TASSEL-GBSv1, 76,1%). . Среди конвейеров, предлагающих высокую точность (> 95%), Fast-GBS вызвал наибольшее количество полиморфизмов (около 35 000 SNP + Indels) и дал самую высокую точность (98,7%). Используя данные последовательности Ion Torrent для тех же 24 строк, мы сравнили производительность Fast-GBS с производительностью TASSEL-GBSv2.Он снова вызвал больше полиморфизмов (25,8 тыс. против 22,9 тыс.), и они оказались более точными (95,2 против 91,1%). Как правило, каталоги SNP, вызываемые из одних и тех же данных секвенирования с использованием разных конвейеров, приводили к сильному перекрытию каталогов SNP (перекрытие 79–92%). Напротив, перекрытие между каталогами SNP, полученными с использованием одного конвейера, но разных технологий секвенирования, было менее значительным (~ 50–70%).
Напротив, перекрытие между каталогами SNP, полученными с использованием одного конвейера, но разных технологий секвенирования, было менее значительным (~ 50–70%).
Образец цитирования: Torkamaneh D, Laroche J, Belzile F (2016) Полногеномный вызов SNP на основе данных генотипирования путем секвенирования (GBS): сравнение семи конвейеров и двух технологий секвенирования.ПЛОС ОДИН 11(8): e0161333. https://doi.org/10.1371/journal.pone.0161333
Редактор: Hector Candela, Universidad Miguel Hernández de Elche, ИСПАНИЯ
Поступила в редакцию: 27 января 2016 г.; Принято: 3 августа 2016 г.; Опубликовано: 22 августа 2016 г.
Авторское право: © 2016 Torkamaneh et al. Это статья с открытым доступом, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.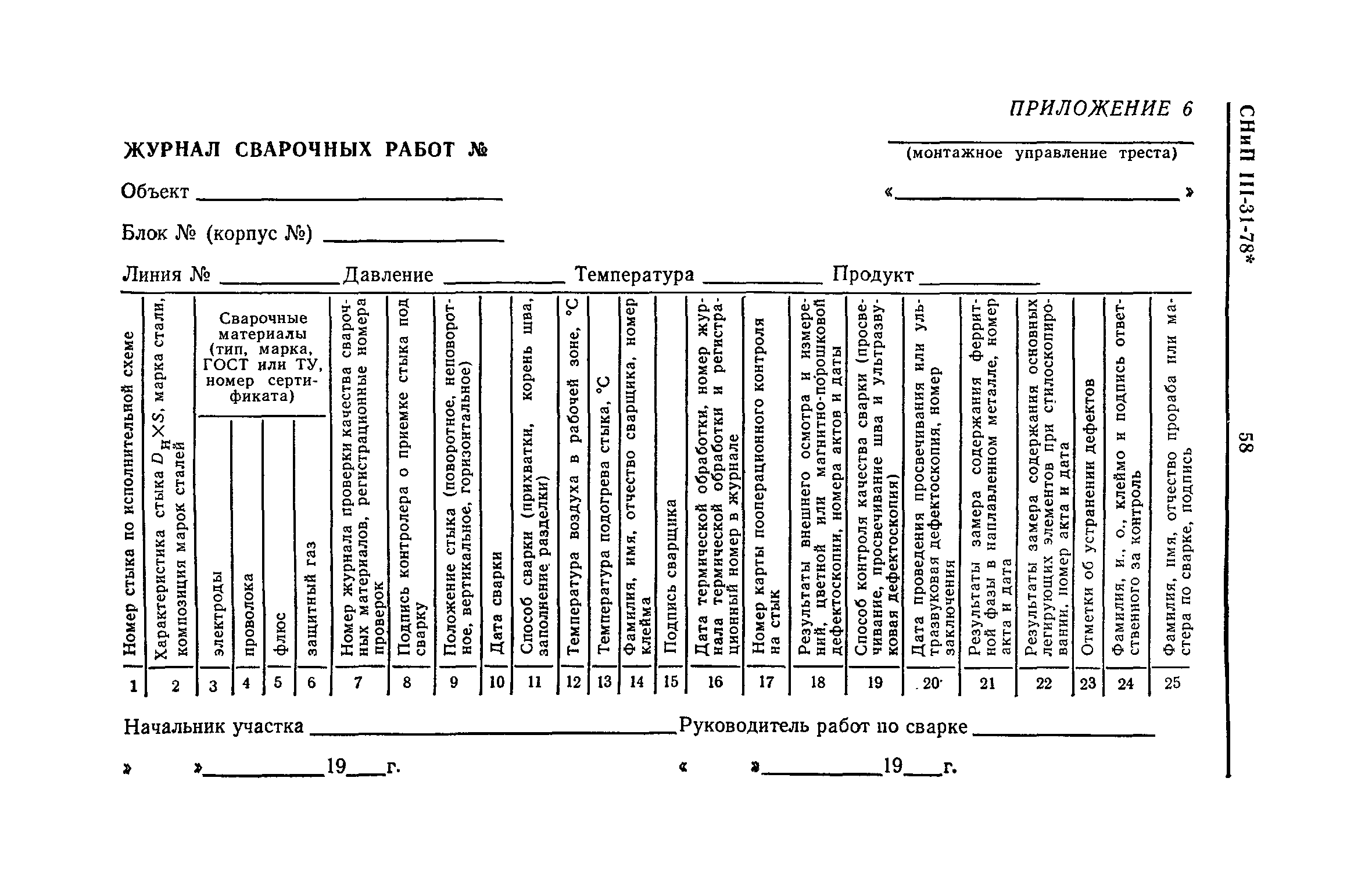
Доступность данных: Все данные (GBS и WGS) доступны в архиве NCBI Sequence Read Archive (SRA) с регистрационным номером SRP# Study, SRP059747 и SRP073237.
Финансирование: Финансирование этого исследования было предоставлено Министерством сельского хозяйства и сельского хозяйства Канады и Канадским альянсом по исследованию полевых культур (грант № AIP-CL23).
Конкурирующие интересы: Авторы заявили об отсутствии конкурирующих интересов.
Введение
Секвенирование следующего поколения (NGS) значительно облегчило разработку методов генотипирования очень большого числа молекулярных маркеров, таких как однонуклеотидные полиморфизмы (SNP).NGS предлагает несколько подходов, которые способны одновременно выполнять обнаружение SNP по всему геному и генотипирование за один шаг, даже у видов, для которых мало или вообще нет генетической информации [1]. Эта революция в открытии генетических маркеров позволяет изучать важные вопросы молекулярной селекции, популяционной генетики, экологической генетики и эволюции.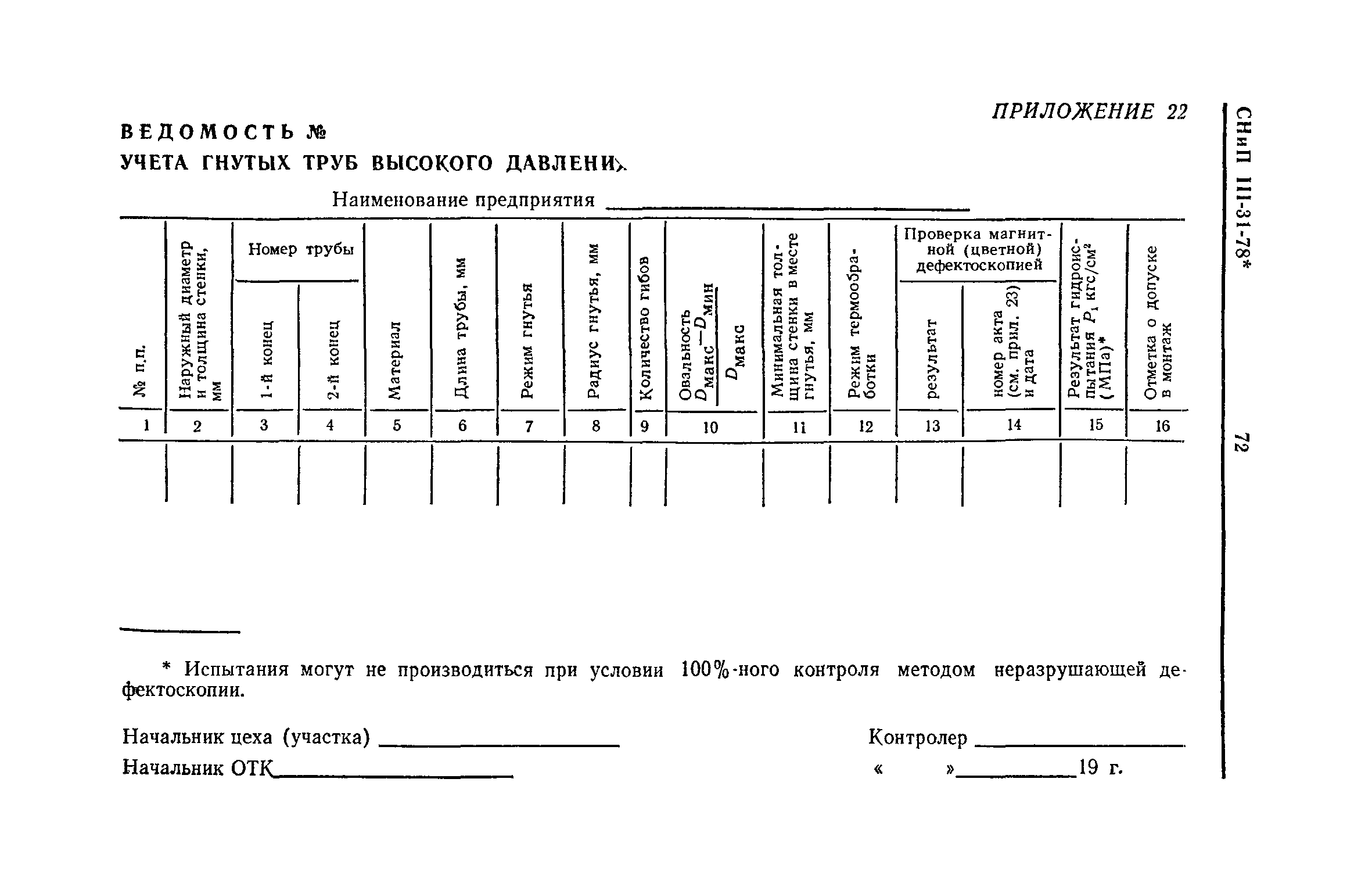 Наиболее широко используемые методы генотипирования, основанные на NGS, используют рестрикционные ферменты для захвата уменьшенного представления генома [2–9].Новые подходы, такие как секвенирование ДНК, связанное с сайтом рестрикции (RAD-seq) и генотипирование посредством секвенирования (GBS), были разработаны в качестве быстрых и надежных подходов к секвенированию мультиплексированных образцов с уменьшенным представлением, которое сочетает в себе обнаружение молекулярных маркеров по всему геному и генотипирование. [1]. Это семейство подходов к генотипированию с уменьшенным представлением, обычно называемое генотипированием путем секвенирования (GBS) [1]. Гибкость и низкая стоимость GBS делают его отличным инструментом для многих приложений и исследовательских вопросов в области генетики и селекции.Такие современные достижения позволяют генотипировать тысячи SNP, при этом вероятность выявления SNP, коррелирующих с интересующими признаками, возрастает [10]. Даже с продвижением NGS для получения миллионов считываний последовательностей за цикл анализ данных для этих новых подходов может быть сложным из-за использования рестрикционных ферментов, мультиплексирования образцов, различной длины фрагмента и переменной глубины считывания [1].
Наиболее широко используемые методы генотипирования, основанные на NGS, используют рестрикционные ферменты для захвата уменьшенного представления генома [2–9].Новые подходы, такие как секвенирование ДНК, связанное с сайтом рестрикции (RAD-seq) и генотипирование посредством секвенирования (GBS), были разработаны в качестве быстрых и надежных подходов к секвенированию мультиплексированных образцов с уменьшенным представлением, которое сочетает в себе обнаружение молекулярных маркеров по всему геному и генотипирование. [1]. Это семейство подходов к генотипированию с уменьшенным представлением, обычно называемое генотипированием путем секвенирования (GBS) [1]. Гибкость и низкая стоимость GBS делают его отличным инструментом для многих приложений и исследовательских вопросов в области генетики и селекции.Такие современные достижения позволяют генотипировать тысячи SNP, при этом вероятность выявления SNP, коррелирующих с интересующими признаками, возрастает [10]. Даже с продвижением NGS для получения миллионов считываний последовательностей за цикл анализ данных для этих новых подходов может быть сложным из-за использования рестрикционных ферментов, мультиплексирования образцов, различной длины фрагмента и переменной глубины считывания [1]. Совершенно очевидно, что конвейеры расширенного анализа стали необходимостью для фильтрации, сортировки и выравнивания этих данных последовательности.Конвейер для GBS должен включать шаги для фильтрации некачественных прочтений, классификации прочтений по пулу или отдельным лицам на основе штрих-кодов последовательности, либо идентификации локусов и аллелей de novo , либо сопоставления прочтений с эталонным геномом индекса для обнаружения полиморфизмов и часто оценка генотипы для каждой особи, включенной в исследование. Как правило, конвейеры для обработки данных GBS делятся на две группы; de novo на основе и на основе ссылок. Когда эталонный геном доступен, чтения из секвенирования с уменьшенным представлением могут быть сопоставлены с эталонным геномом, а SNP могут быть вызваны как для проектов ресеквенирования всего генома [11-12].К настоящему времени было разработано несколько конвейеров анализа GBS на основе эталонов. Наиболее широко используемыми конвейерами анализа GBS на основе эталонов являются: TASSEL-GBS (v1 и v2), Stacks, IGST и Fast-GBS (самый последний конвейер, Torkamaneh et al.
Совершенно очевидно, что конвейеры расширенного анализа стали необходимостью для фильтрации, сортировки и выравнивания этих данных последовательности.Конвейер для GBS должен включать шаги для фильтрации некачественных прочтений, классификации прочтений по пулу или отдельным лицам на основе штрих-кодов последовательности, либо идентификации локусов и аллелей de novo , либо сопоставления прочтений с эталонным геномом индекса для обнаружения полиморфизмов и часто оценка генотипы для каждой особи, включенной в исследование. Как правило, конвейеры для обработки данных GBS делятся на две группы; de novo на основе и на основе ссылок. Когда эталонный геном доступен, чтения из секвенирования с уменьшенным представлением могут быть сопоставлены с эталонным геномом, а SNP могут быть вызваны как для проектов ресеквенирования всего генома [11-12].К настоящему времени было разработано несколько конвейеров анализа GBS на основе эталонов. Наиболее широко используемыми конвейерами анализа GBS на основе эталонов являются: TASSEL-GBS (v1 и v2), Stacks, IGST и Fast-GBS (самый последний конвейер, Torkamaneh et al. (неопубликованный)) [9, 13–15]. . В отсутствие эталонного генома необходимо идентифицировать пары почти идентичных прочтений (предположительно представляющих альтернативные аллели локуса). Наиболее часто используемыми конвейерами для такого подхода на основе de novo являются UNEAK и Stacks [15, 16].
(неопубликованный)) [9, 13–15]. . В отсутствие эталонного генома необходимо идентифицировать пары почти идентичных прочтений (предположительно представляющих альтернативные аллели локуса). Наиболее часто используемыми конвейерами для такого подхода на основе de novo являются UNEAK и Stacks [15, 16].
Наконец, в настоящее время доступны различные платформы секвенирования NGS, предлагающие различные преимущества. Например, в то время как технология Illumina предлагает очень высокую пропускную способность и качество чтения, это обычно достигается за счет скорости, поскольку для завершения цикла требуется около двух недель. Напротив, технология Ion Torrent [17] предлагает большую скорость (4 часа) за счет более низкой пропускной способности и качества чтения. В зависимости от ограничений та или иная технология может оказаться более подходящей.В идеале хотелось бы, чтобы конвейеры вызовов SNP одинаково хорошо работали с обоими типами считываемых данных.
В этом исследовании мы всесторонне сравнили существующие конвейеры анализа GBS на основе количества вызванных SNP, точности полученных генотипов, а также скорости и простоты использования этих конвейеров. Мы также сравнили результаты, полученные с помощью ридов Illumina и Ion Torrent. Наконец, мы изучили количество перекрытий в локусах SNP, которые вызывались с использованием разных конвейеров.
Мы также сравнили результаты, полученные с помощью ридов Illumina и Ion Torrent. Наконец, мы изучили количество перекрытий в локусах SNP, которые вызывались с использованием разных конвейеров.
Материалы и методы
Образцы и платформа для секвенирования
Соя ( Glycine max L .) — диплоидный вид с 20 парами хромосом и геномом среднего размера (1,1 Гб). Поскольку это автогамный вид, линии/сорта сои размножаются правильно и в высокой степени гомозиготны. Набор из 23 канадских линий сои и одной интродукции растений (PI) был подвергнут анализу GBS. Эти же линии были повторно секвенированы, как описано ранее Torkamaneh и Belzile [18].Используя одну и ту же ДНК, две библиотеки GBS были созданы после расщепления Ape KI: одна для секвенирования Illumina (согласно Elshire et al. [6]), а другая для секвенирования Ion Torrent (согласно Mascher et al. [19]). . Одностороннее секвенирование проводили либо на Illumina HiSeq 2000 в Инновационном центре McGill University-Génome Québec в Монреале, Канада, либо на машине Ion Proton в Институте интегративной и системной биологии (IBIS) Университета Лаваля, Квебек. Канада. Всего на платформе Illumina было сгенерировано 42 миллиона прочтений размером 100 п.н., а на платформе Ion Torrent — 38 миллионов прочтений размером от 50 до 135 п.н.Все данные (GBS и WGS) доступны в архиве NCBI Sequence Read Archive (SRA) под номерами SRP059747 (последовательности Illumina) и SRP073237 (последовательности Ion Torrent).
Канада. Всего на платформе Illumina было сгенерировано 42 миллиона прочтений размером 100 п.н., а на платформе Ion Torrent — 38 миллионов прочтений размером от 50 до 135 п.н.Все данные (GBS и WGS) доступны в архиве NCBI Sequence Read Archive (SRA) под номерами SRP059747 (последовательности Illumina) и SRP073237 (последовательности Ion Torrent).
Трубопроводы анализа GBS
Мы использовали два вызывающих варианта de novo и пять эталонных конвейеров (эталонный геном Williams82; [20]) для вызова SNP. Мы запускали все пайплайны в одинаковых условиях глубины охвата (minDP≥2), максимального несоответствия для выравнивания (n = 3), максимального количества отсутствующих данных (MaxMD = 80%) и минимальной частоты минорных аллелей (MinMAF≥0.05). Ниже мы кратко опишем процессы для каждого пайплайна. Для вычислений мы использовали систему Linux с 10 ЦП и 25 ГБ памяти. В дополнение к описаниям, представленным ниже, в таблице S1 приведена сводка различных компонентов каждого конвейера, и мы предоставляем все командные строки, используемые в этой работе, в качестве вспомогательной информации (текст S1).
Fast-GBS.
Конвейер анализа Fast-GBS был разработан путем интеграции общедоступных пакетов с инструментами собственной разработки.Основные функции включают в себя: (1) демультиплексирование и очистку считываний необработанных последовательностей; (2) оценка качества чтения и составление карт; (3) фильтрация сопоставленных прочтений и оценка сложности библиотеки; (4) перегруппировка и построение локальных гаплотипов; (5) подходящие популяционные частоты и отдельные гаплотипы; (5) необработанный вариант вызова; (6) вариантная и индивидуальная фильтрация; (7) идентификация высококонсистентных вариантов. Поскольку исследователи не всегда могут иметь немедленный доступ к ресурсам кластера, этот конвейер позволяет либо параллельную обработку большого количества образцов в кластере, либо последовательную обработку нескольких образцов на одной машине.
IGST (инструмент IBIS для генотипирования посредством секвенирования).
Конвейер, реализованный на языке программирования Perl, был разработан для обработки данных чтения последовательностей Illumina. Шаги, задействованные в конвейере, выполнялись в отдельных сценариях оболочки. В этом конвейере используются различные общедоступные программные инструменты (набор инструментов FASTX, BWA, SAMtools, VCFtools), а также некоторые собственные инструменты [11, 21, 22]. Полученные необработанные SNP были дополнительно отфильтрованы с использованием VCFtools на основе глубины чтения, отсутствующих данных в генотипах и частоте минорных аллелей.Гетерозиготная коррекция выполняется собственным скриптом Python.
Шаги, задействованные в конвейере, выполнялись в отдельных сценариях оболочки. В этом конвейере используются различные общедоступные программные инструменты (набор инструментов FASTX, BWA, SAMtools, VCFtools), а также некоторые собственные инструменты [11, 21, 22]. Полученные необработанные SNP были дополнительно отфильтрованы с использованием VCFtools на основе глубины чтения, отсутствующих данных в генотипах и частоте минорных аллелей.Гетерозиготная коррекция выполняется собственным скриптом Python.
TASSEL-GBS (версия 1 и 2).
Конвейеры TASSEL-GBS реализованы на языке программирования Java. В настоящее время доступны две версии: TASSEL-GBS v1 (TASSEL 3.0) [13] и TASSEL-GBS v2 (TASSEL 5.0) [14]. Оба конвейера работают одинаково и требуют, чтобы все операции чтения были урезаны до одинаковой длины (64 п.н. в версии 1, до 92 п.н. в версии 2), а идентичные операции чтения свернуты в теги. Эти теги затем выравниваются по отношению к эталонному геному, и из выровненных тегов вызываются SNP.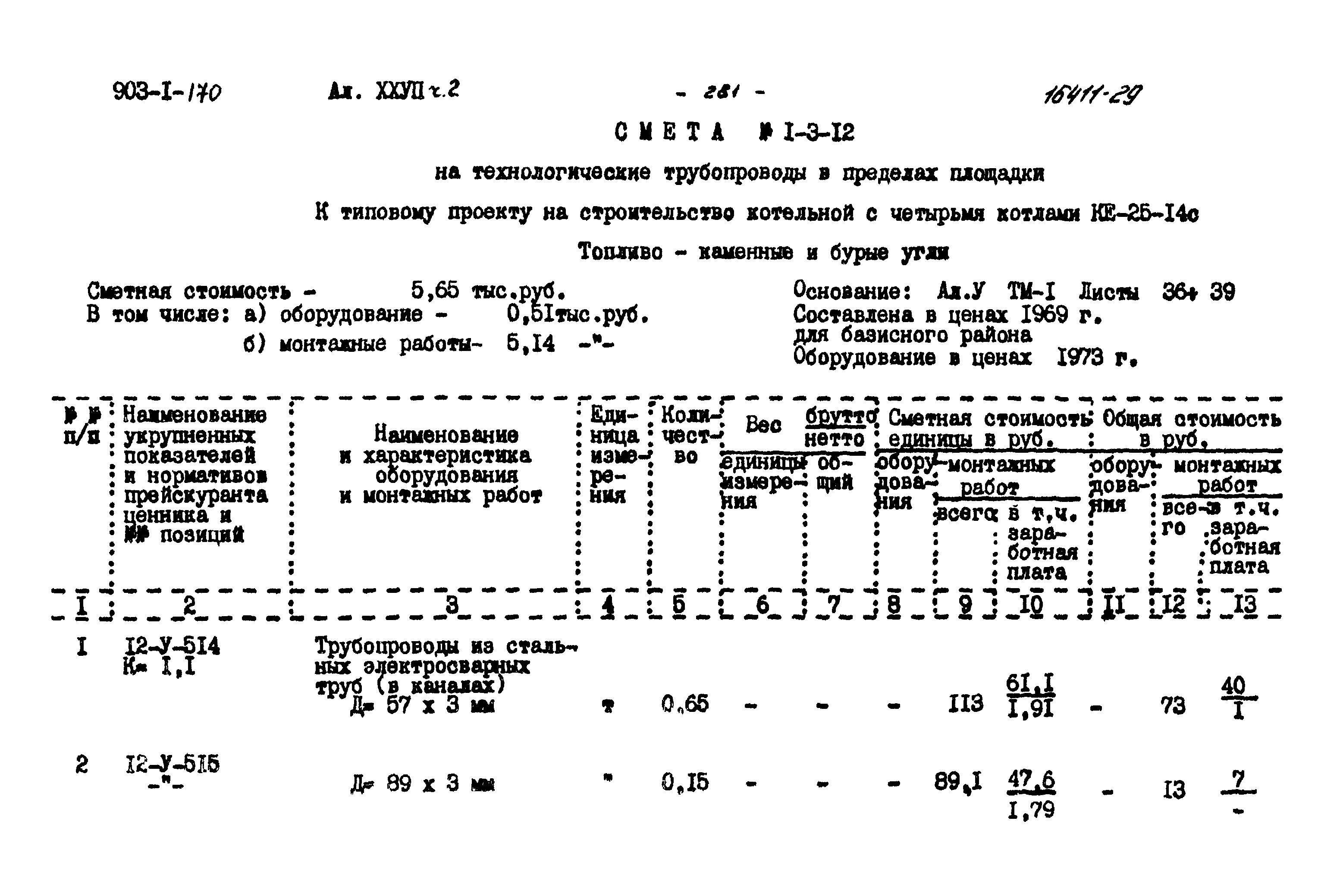 Основными изменениями, реализованными в TASSEL-GBS v2, являются: 1) возможность использования более длинных меток для повышения точности выравнивания с эталонным геномом и 2) расширенный этап обнаружения и производства SNP.
Основными изменениями, реализованными в TASSEL-GBS v2, являются: 1) возможность использования более длинных меток для повышения точности выравнивания с эталонным геномом и 2) расширенный этап обнаружения и производства SNP.
UNEAK (Универсальный сетевой аналитический комплект).
Общий дизайн UNEAK выглядит следующим образом: 1) чтения обрезаются до 64 п.н.; 2) идентичные риды длиной 64 п.н. свернуты в теги; 3) попарное выравнивание идентифицирует пары тегов, имеющие несовпадение одной пары оснований. Эти несовпадения в одной паре оснований являются кандидатами в SNP.«Сетевой фильтр» используется для отбрасывания повторов, паралогов и ошибок секвенирования, что приводит к набору взаимных пар тегов или SNP.
Стеки(на основе эталона и
de novo ). Необработанные входные данные для Stacks представляют собой секвенированные фрагменты ДНК из любого протокола GBS на основе рестрикционных ферментов. Стеки могут обрабатывать необработанные данные секвенирования для идентификации локусов de novo или путем выравнивания с эталонным геномом [10]. Независимо от того, собраны ли данные de novo или сопоставлены с эталонным геномом, многие последующие шаги в стеках являются общими.Конвейер можно описать следующим образом: (1) Необработанные чтения последовательностей демультиплексируются и очищаются (process_radtags). (2) Данные от каждого индивидуума группируются в локусы и идентифицируются сайты полиморфных нуклеотидов (ustacks или pstacks для невыровненных или выровненных данных соответственно). (3) Локусы группируются по отдельным лицам, и составляется каталог (cstacks). (4) Локусы от каждого индивидуума сопоставляются с каталогом для определения аллельного состояния в каждом локусе у каждого индивидуума (sstacks).(5) Аллельные состояния либо преобразуются в набор картируемых генотипов (для генетической карты) с использованием генотипов, либо подвергаются популяционно-генетической статистике через популяции с записью результатов в один или несколько выходных файлов.
Независимо от того, собраны ли данные de novo или сопоставлены с эталонным геномом, многие последующие шаги в стеках являются общими.Конвейер можно описать следующим образом: (1) Необработанные чтения последовательностей демультиплексируются и очищаются (process_radtags). (2) Данные от каждого индивидуума группируются в локусы и идентифицируются сайты полиморфных нуклеотидов (ustacks или pstacks для невыровненных или выровненных данных соответственно). (3) Локусы группируются по отдельным лицам, и составляется каталог (cstacks). (4) Локусы от каждого индивидуума сопоставляются с каталогом для определения аллельного состояния в каждом локусе у каждого индивидуума (sstacks).(5) Аллельные состояния либо преобразуются в набор картируемых генотипов (для генетической карты) с использованием генотипов, либо подвергаются популяционно-генетической статистике через популяции с записью результатов в один или несколько выходных файлов.
Точность генотипа
Для оценки точности вызовов генотипов мы использовали собственный сценарий для сравнения генотипов, названных с использованием GBS, с генотипами, названными в тех же локусах после WGS. Секвенирование и определение SNP в этой коллекции из 24 линий сои ранее было описано Torkamaneh и Belzile [18].Вкратце, линии сои были секвенированы до средней глубины охвата 9x, и было достигнуто покрытие генома 96%. Чтения парных концов Illumina были сопоставлены с эталонным геномом сои (Williams82) с использованием BWA, а генотипы в полиморфных локусах были названы с помощью SAMtools. Варианты с двумя или более альтернативными аллелями были удалены. Таким образом, среди этих линий было названо в общей сложности 3,6 млн SNP. В качестве дополнительного средства для измерения качества генотипа мы оценили долю отсутствующих данных и гетерозиготных вызовов, полученных с каждым конвейером анализа.Для конвейеров de novo мы сопоставили теги, поддерживающие SNP, с эталонным геномом, чтобы найти физическое положение, а затем сравнили их с набором данных WGS.
Секвенирование и определение SNP в этой коллекции из 24 линий сои ранее было описано Torkamaneh и Belzile [18].Вкратце, линии сои были секвенированы до средней глубины охвата 9x, и было достигнуто покрытие генома 96%. Чтения парных концов Illumina были сопоставлены с эталонным геномом сои (Williams82) с использованием BWA, а генотипы в полиморфных локусах были названы с помощью SAMtools. Варианты с двумя или более альтернативными аллелями были удалены. Таким образом, среди этих линий было названо в общей сложности 3,6 млн SNP. В качестве дополнительного средства для измерения качества генотипа мы оценили долю отсутствующих данных и гетерозиготных вызовов, полученных с каждым конвейером анализа.Для конвейеров de novo мы сопоставили теги, поддерживающие SNP, с эталонным геномом, чтобы найти физическое положение, а затем сравнили их с набором данных WGS.
Результаты
Вариант вызова с разными конвейерами с использованием данных чтения Illumina
Чтобы оценить эффективность различных конвейеров анализа GBS, мы проанализировали общедоступные данные GBS (прочтения Illumina 100 п. н.) из набора из 24 ранее изученных линий сои. Мы сравнили пять эталонных конвейеров анализа: TASSEL-GBS v1 и v2, Stacks, IGST и Fast-GBS.Мы также сравнили два широко используемых вызывающих абонента de novo : UNEAK и Stacks. Мы использовали одинаковое количество чтений для всех анализов (42M чтений) и попытались подобрать параметры, максимально схожие для всех пайплайнов (подробности см. в M&M). Как показано в Таблице 1, большие различия в количестве вызванных SNP наблюдались как для de novo , так и для эталонных конвейеров. Среди первых Стэкс назвал наименьшее количество SNP, примерно в 2 раза меньше, чем UNEAK (13 303 против 24 743).Количество SNP, вызванных UNEAK, было ненамного ниже среднего количества SNP, вызванных референсными конвейерами (32 423). Среди эталонных конвейеров количество вызванных SNP варьировалось от 18 941 (стеки) до 54 412 (TASSEL-GBS v1), т. е. разница в 2,8 раза. Три других референсных пайплайна были намного ближе к среднему значению, вызывая примерно от 25 до 35 тысяч SNP.
н.) из набора из 24 ранее изученных линий сои. Мы сравнили пять эталонных конвейеров анализа: TASSEL-GBS v1 и v2, Stacks, IGST и Fast-GBS.Мы также сравнили два широко используемых вызывающих абонента de novo : UNEAK и Stacks. Мы использовали одинаковое количество чтений для всех анализов (42M чтений) и попытались подобрать параметры, максимально схожие для всех пайплайнов (подробности см. в M&M). Как показано в Таблице 1, большие различия в количестве вызванных SNP наблюдались как для de novo , так и для эталонных конвейеров. Среди первых Стэкс назвал наименьшее количество SNP, примерно в 2 раза меньше, чем UNEAK (13 303 против 24 743).Количество SNP, вызванных UNEAK, было ненамного ниже среднего количества SNP, вызванных референсными конвейерами (32 423). Среди эталонных конвейеров количество вызванных SNP варьировалось от 18 941 (стеки) до 54 412 (TASSEL-GBS v1), т. е. разница в 2,8 раза. Три других референсных пайплайна были намного ближе к среднему значению, вызывая примерно от 25 до 35 тысяч SNP.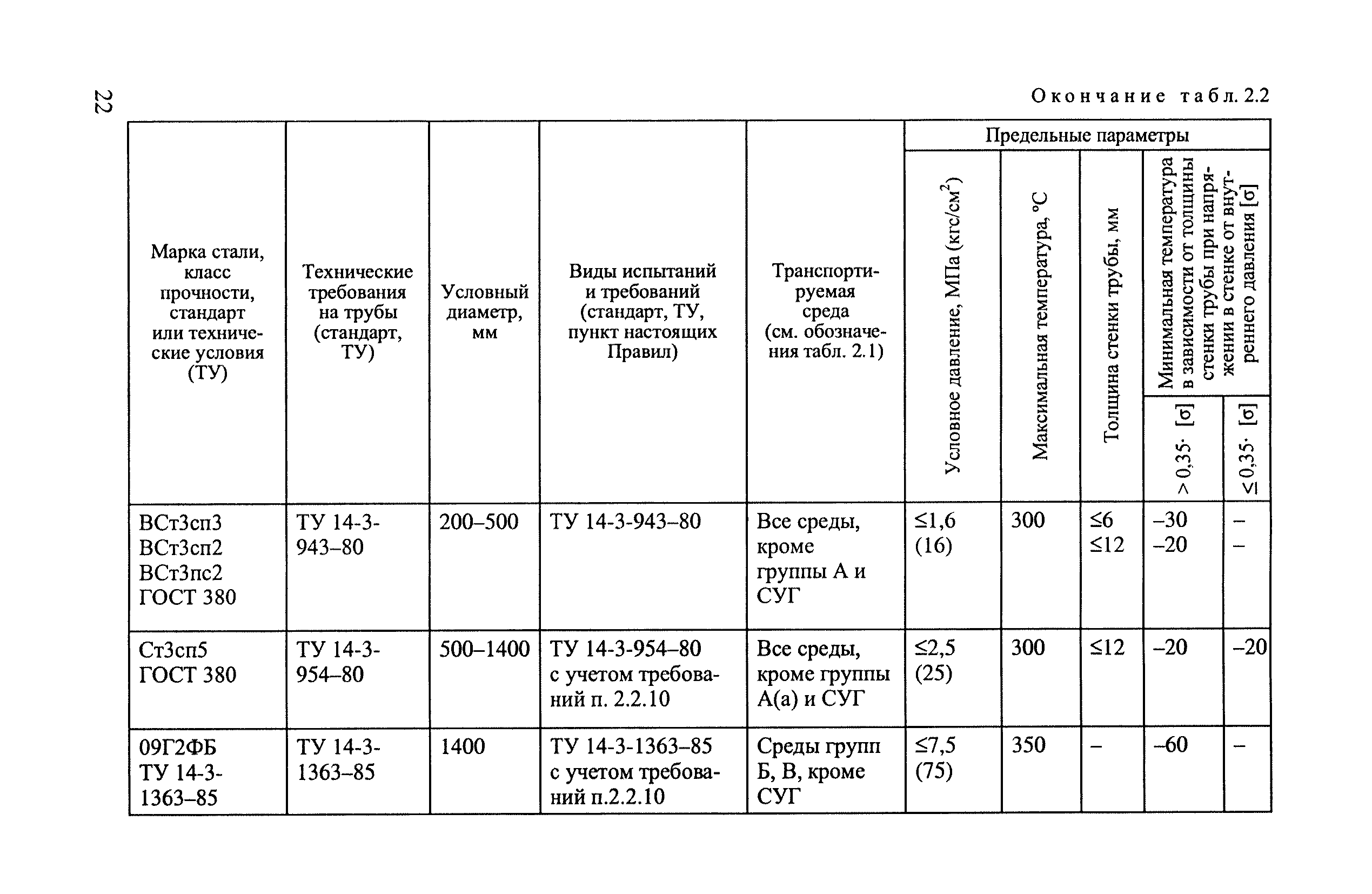 Помимо вызова SNP, IGST и Fast-GBS также могли вызывать вставки. В обоих случаях они внесли дополнительные 12–13% в общее количество вариантов.
Помимо вызова SNP, IGST и Fast-GBS также могли вызывать вставки. В обоих случаях они внесли дополнительные 12–13% в общее количество вариантов.
Таблица 1. Количество SNP и вставок, обнаруженных среди 24 линий сои с использованием семи различных конвейеров биоинформатики в считываниях Illumina.
Также указываются время и объем памяти, необходимые для запуска каждого конвейера.
https://doi.org/10.1371/journal.pone.0161333.t001
Fast-GBS и TASSEL-GBS v1 оказались самыми быстрыми среди эталонных конвейеров (~1 час 55 минут), тогда как IGST оказался самым медленным, для завершения анализа требуется почти 13 часов.Среди конвейеров de novo UNEAK был почти в три раза быстрее, чем Stacks (1 ч 21 мин против 3 ч 07 мин) и оказался самым быстрым из всех конвейеров. С точки зрения требуемой памяти здесь также наблюдались очень большие различия. Среди конвейеров de novo для UNEAK требовалось почти в три раза больше места на диске по сравнению со стеками (20 Гб против 7 Гб). Среди эталонных конвейеров различия были еще больше, поскольку для IGST требовалось в 17,1 раза больше памяти (240 Гб), чем для стеков (14 Гб).
Среди эталонных конвейеров различия были еще больше, поскольку для IGST требовалось в 17,1 раза больше памяти (240 Гб), чем для стеков (14 Гб).
Точность и эффективность конвейеров биоинформатики GBS
Чтобы проверить качество данных SNP, полученных с использованием эталонных конвейеров, мы сначала измерили количество отсутствующих данных, а затем оценили точность генотипа, сравнив генотипы, полученные с помощью GBS, с истинными генотипами, обнаруженными в результате полногеномного повторного секвенирования тех же линий. .Оценки точности SNP, называемых GBS, были выполнены для всех SNP для всех пайплайнов при одинаковых уровнях допуска отсутствующих данных (≤80%) и частоте минорных аллелей (≥0,05). Как видно из таблицы 2, среди эталонных пайплайнов доля отсутствующих данных варьировалась от 28 % (TASSEL GBS v1) до 57,3 % (стеки). Среди конвейеров de novo доля отсутствующих данных была менее изменчивой: от 39,4% (стеки) до 41,3% (UNEAK).
Когда мы сравнили генотипы, полученные с помощью каждого конвейера, с генотипами, полученными в результате повторного секвенирования, мы обнаружили, что 98.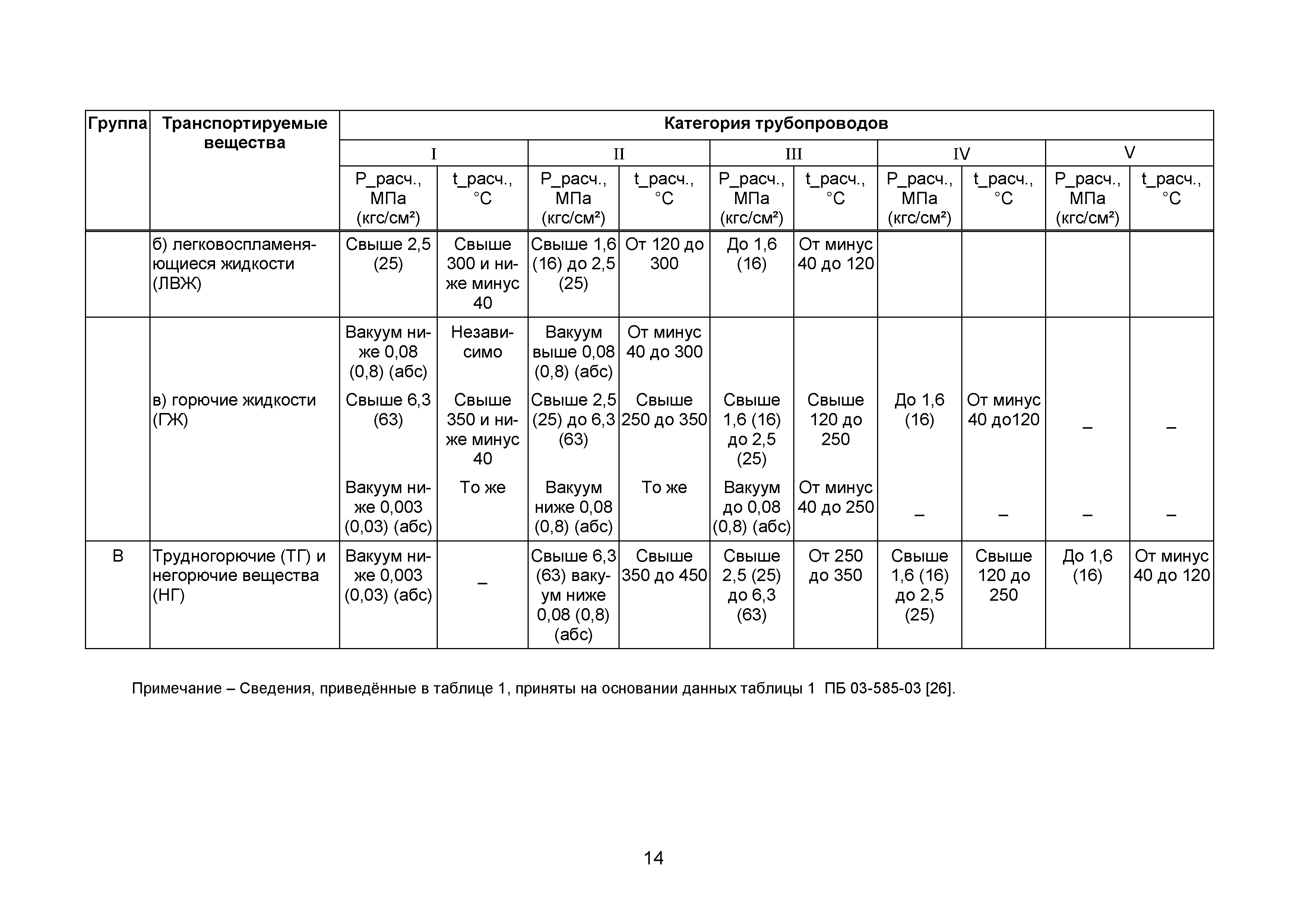 7% генотипов SNP, вызванных с использованием конвейера Fast-GBS, совпали с истинными генотипами. Подобные уровни точности были обнаружены для SNP, вызванных с помощью IGST (98,4%). За единственным исключением, все эталонные пайплайны достигли уровня точности >92%. TASSEL-GBS v1 оказался наименее точным из этих конвейеров, поскольку только 76,1% названных им генотипов были идентичны данным повторного секвенирования. Среди конвейеров de novo точность вызовов генотипов была лишь немного ниже (в среднем 93,7%), чем точность, полученная с эталонными конвейерами, отличными от TASSEL-GBS v1 (95.в среднем 6%).
7% генотипов SNP, вызванных с использованием конвейера Fast-GBS, совпали с истинными генотипами. Подобные уровни точности были обнаружены для SNP, вызванных с помощью IGST (98,4%). За единственным исключением, все эталонные пайплайны достигли уровня точности >92%. TASSEL-GBS v1 оказался наименее точным из этих конвейеров, поскольку только 76,1% названных им генотипов были идентичны данным повторного секвенирования. Среди конвейеров de novo точность вызовов генотипов была лишь немного ниже (в среднем 93,7%), чем точность, полученная с эталонными конвейерами, отличными от TASSEL-GBS v1 (95.в среднем 6%).
Среди растений недавние или древние события полиплоидизации могут генерировать паралоги, которые могут быть ошибочно приняты за представление аллелей одного локуса на основании чтения коротких последовательностей. Поэтому мы исследовали как общее количество гетерозиготных вызовов генотипа, так и количество локусов, содержащих большую долю (> 50%) гетерозиготных вызовов. Как видно из таблицы 2, пайплайны de novo вызвали аналогичную долю гетерозиготных генотипов (~3,7 и 5,3% для Stacks и UNEAK соответственно) и не сохранили ни одного локуса с большой долей гетерозигот.Среди эталонных пайплайнов Fast-GBS и TASSEL-GBS v1 назвали наименьшее и самое гетерозиготное генотипы (3,4 и 11,5% соответственно). Кроме того, TASSEL-GBS v1 назвал наибольшее количество локусов с большой долей гетерозиготных генотипов (1125), в то время как Stacks назвал только 65 локусов с более чем 50% гетерозигот.
Как видно из таблицы 2, пайплайны de novo вызвали аналогичную долю гетерозиготных генотипов (~3,7 и 5,3% для Stacks и UNEAK соответственно) и не сохранили ни одного локуса с большой долей гетерозигот.Среди эталонных пайплайнов Fast-GBS и TASSEL-GBS v1 назвали наименьшее и самое гетерозиготное генотипы (3,4 и 11,5% соответственно). Кроме того, TASSEL-GBS v1 назвал наибольшее количество локусов с большой долей гетерозиготных генотипов (1125), в то время как Stacks назвал только 65 локусов с более чем 50% гетерозигот.
Перекрытие между каталогами SNP
Затем мы определили степень перекрытия каталогов SNP, полученных с использованием различных конвейеров, и их точность.Мы выбрали Fast-GBS в качестве основы для сравнения из-за его способности очень точно вызывать большое количество SNP. Как показано в таблице 3, среди эталонных конвейеров наибольшее совпадение наблюдалось между Fast-GBS и Stacks (> 96%), а 92% SNP, вызываемых с помощью IGST, также были обнаружены в наборе данных Fast-GBS. Напротив, TASSEL-GBS v1 показал самое низкое перекрытие (36,7%) с Fast-GBS. Конвейеры de novo показали аналогичные уровни перекрытия с Fast-GBS (стеки = 89,1% и UNEAK = 87.5%). В дополнительном анализе (не показанном в таблице 3) мы измерили перекрытие между двумя конвейерами de novo ; около 67% SNP, вызванных Stacks, также были обнаружены в наборе данных UNEAK. Таким образом, эти два конвейера de novo , по-видимому, идентифицируют довольно разные подмножества более обширного каталога SNP, полученного с использованием Fast-GBS.
Напротив, TASSEL-GBS v1 показал самое низкое перекрытие (36,7%) с Fast-GBS. Конвейеры de novo показали аналогичные уровни перекрытия с Fast-GBS (стеки = 89,1% и UNEAK = 87.5%). В дополнительном анализе (не показанном в таблице 3) мы измерили перекрытие между двумя конвейерами de novo ; около 67% SNP, вызванных Stacks, также были обнаружены в наборе данных UNEAK. Таким образом, эти два конвейера de novo , по-видимому, идентифицируют довольно разные подмножества более обширного каталога SNP, полученного с использованием Fast-GBS.
Чтобы лучше понять генотипическую точность среди различных подмножеств общих или уникальных SNP, мы подготовили две отдельные диаграммы Венна, каждая из которых включает только четыре конвейера (для ясности), с Fast-GBS, включенным в обе панели (рис. 1).Что выделяется на этом рисунке, так это то, что SNP, вызываемые более чем одним конвейером, обычно были очень точными (средневзвешенная точность = 94,8%). Напротив, за единственным исключением Fast-GBS, SNP, вызываемые одним конвейером, обычно были гораздо менее точными (средневзвешенная точность = 66,3%). Наиболее поразительно отметим, что TASSEL-GBS v1 назвал очень большое количество уникальных SNP (более 30 000), которые показывают низкую точность (65%). Уникальные SNP, вызываемые другими пайплайнами, также обычно демонстрировали низкую точность, но их было гораздо меньше, и поэтому в целом они оказывали меньшее влияние.
Наиболее поразительно отметим, что TASSEL-GBS v1 назвал очень большое количество уникальных SNP (более 30 000), которые показывают низкую точность (65%). Уникальные SNP, вызываемые другими пайплайнами, также обычно демонстрировали низкую точность, но их было гораздо меньше, и поэтому в целом они оказывали меньшее влияние.
Рис. 1. Диаграмма Венна, представляющая степень перекрытия между локусами SNP, вызванными с использованием семи конвейеров биоинформатики.
Проценты показывают предполагаемую точность для всех групп SNP (уникальных или общих).
https://doi.org/10.1371/journal.pone.0161333.g001
Причины плохой работы некоторых конвейеров
Учитывая наблюдаемую разницу в количестве вызываемых SNP и их точности, мы решили исследовать причины ошибочных вызовов.Для проведения этого исследования мы использовали систематический подход, показанный на рис. 2. Мы разделили каталог SNP на две категории, точные и неточные, на основе сравнения вызовов, полученных с помощью GBS, и вызовов, полученных в результате WGS. Затем неточные SNP были классифицированы либо как уникальные для одного конвейера, либо как общие как минимум для двух конвейеров. Чтобы исследовать уникальные «слабые места» пайплайнов, мы сосредоточили наше внимание на уникальных неточных SNP. Первым шагом в этом исследовании было классифицировать эти неточные SNP как поддерживаемые считываниями, отображаемыми в уникальное положение в геноме, или считываниями, отображаемыми в нескольких позициях.В первом случае ошибки генотипирования были приписаны ошибке вызывающим вариант (например, из-за ошибок секвенирования или амплификации ПЦР). Во втором случае мы пришли к выводу, что картирование прочтений более чем в одном месте генома может быть результатом этих прочтений, происходящих либо из паралогов, либо из повторяющихся областей. Чтобы решить эту проблему, мы сопоставили чтения с замаскированным эталонным геномом (Phytozome V9: Gmax-189-hardmasked.fa), чтобы оценить долю неточных SNP, происходящих из повторяющихся областей.SNP, которые больше не присутствовали в каталоге, полученном в результате картирования замаскированного эталонного генома, считались связанными с повторяющимися последовательностями.
Затем неточные SNP были классифицированы либо как уникальные для одного конвейера, либо как общие как минимум для двух конвейеров. Чтобы исследовать уникальные «слабые места» пайплайнов, мы сосредоточили наше внимание на уникальных неточных SNP. Первым шагом в этом исследовании было классифицировать эти неточные SNP как поддерживаемые считываниями, отображаемыми в уникальное положение в геноме, или считываниями, отображаемыми в нескольких позициях.В первом случае ошибки генотипирования были приписаны ошибке вызывающим вариант (например, из-за ошибок секвенирования или амплификации ПЦР). Во втором случае мы пришли к выводу, что картирование прочтений более чем в одном месте генома может быть результатом этих прочтений, происходящих либо из паралогов, либо из повторяющихся областей. Чтобы решить эту проблему, мы сопоставили чтения с замаскированным эталонным геномом (Phytozome V9: Gmax-189-hardmasked.fa), чтобы оценить долю неточных SNP, происходящих из повторяющихся областей.SNP, которые больше не присутствовали в каталоге, полученном в результате картирования замаскированного эталонного генома, считались связанными с повторяющимися последовательностями. Оставшиеся прочтения, которые успешно сопоставили с несколькими сайтами в замаскированном эталонном геноме, были проанализированы с помощью поиска BLAST для обнаружения паралогии. Чтение считалось производным от паралога, когда мы встречали как минимум 2 совпадения со 100% охватом и минимум 96% идентичности. В среднем считывания, происходящие из паралогичных локусов (как определено выше), имели 2.4 попадания в геном.
Оставшиеся прочтения, которые успешно сопоставили с несколькими сайтами в замаскированном эталонном геноме, были проанализированы с помощью поиска BLAST для обнаружения паралогии. Чтение считалось производным от паралога, когда мы встречали как минимум 2 совпадения со 100% охватом и минимум 96% идентичности. В среднем считывания, происходящие из паралогичных локусов (как определено выше), имели 2.4 попадания в геном.
Результаты этого анализа показаны в Таблице 4. Поскольку большинство пайплайнов предоставили в основном точный (>92%) набор SNP, каждый пайплайн вызывал лишь несколько сотен уникальных неточных SNP, за единственным исключением TASSEL-GBS v1 ( 9828 уникальных неточных SNP). Меньшая часть (от 11,5 до 29,7%) уникальных неточных SNP была подтверждена картированием ридов в одну позицию в геноме и считалась результатом ошибки в вызове вариантов. Большинство (от 70,3 до 88.5%) неточных SNP были подтверждены картированием прочтений более чем с одной областью генома. Среди них подавляющее большинство было связано с картированием прочтений в паралогические области (от 74 до 93%).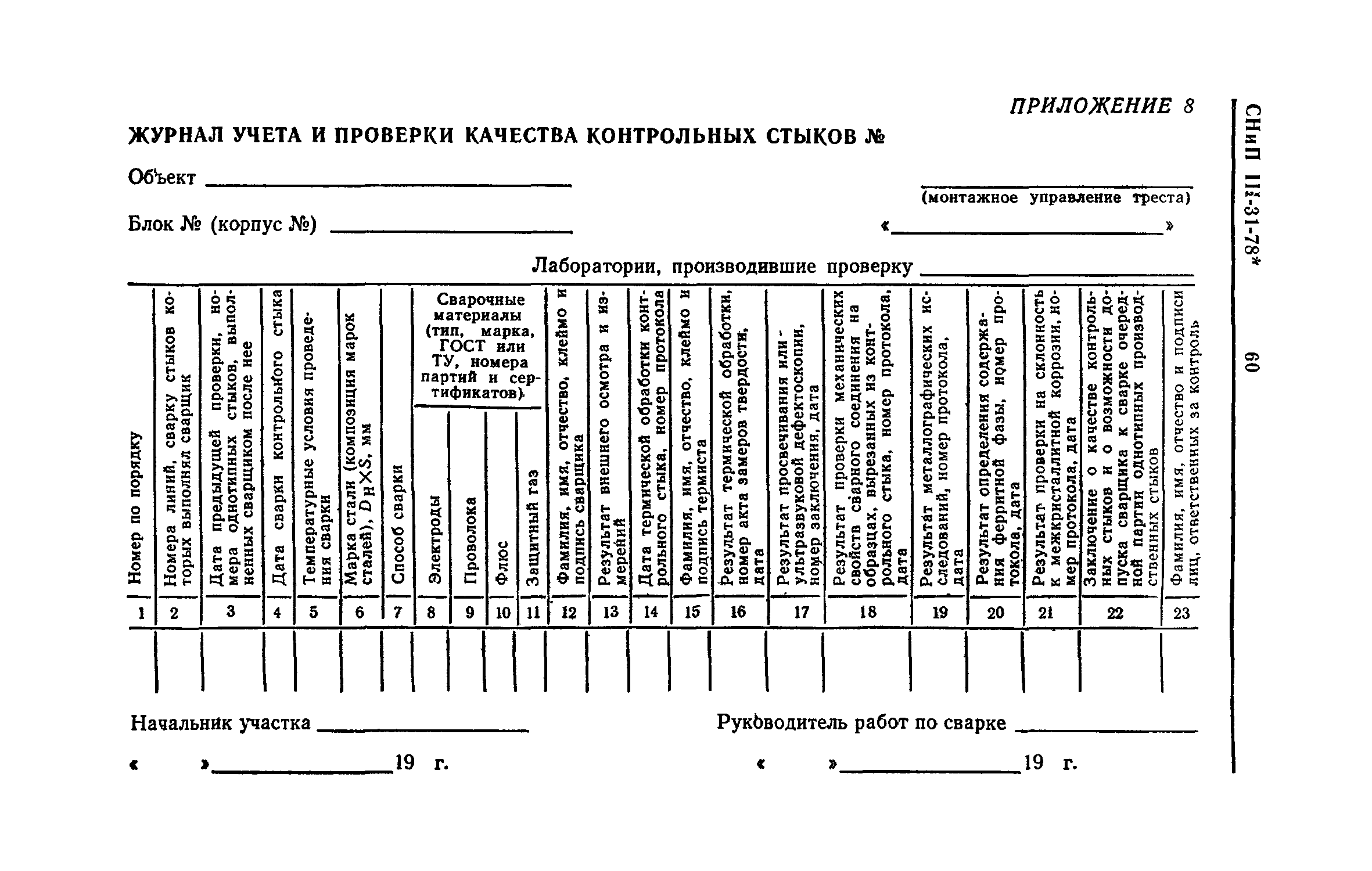 Таким образом, мы пришли к выводу, что большинство ошибок генотипирования сои можно отнести к присутствию паралогов, и что TASSEL-GBS v1 оказался, безусловно, конвейером, наиболее подверженным ошибочным вызовам из-за этого.
Таким образом, мы пришли к выводу, что большинство ошибок генотипирования сои можно отнести к присутствию паралогов, и что TASSEL-GBS v1 оказался, безусловно, конвейером, наиболее подверженным ошибочным вызовам из-за этого.
Другим результатом, который потребовал исследования, было относительно небольшое количество SNP, вызванных Stacks, поскольку как de novo , так и эталонные версии Stacks вызывали наименьшее количество SNP.Мы исследовали эффективность шага демультиплексирования, поскольку он уже был описан как проблематичный. В нашем анализе мы обнаружили, что 19,7% считываний Illumina не удалось присвоить конкретному файлу штрих-кода, что намного выше, чем у других конвейеров. Чтобы измерить влияние такого уменьшения количества чтений, доступных для вызова SNP, мы использовали альтернативный инструмент демультиплексирования (Sabre) вместо того, который предоставляется в Stacks. Доля пропущенных прочтений уменьшилась до ~ 2%, а количество SNP, вызванных с использованием этого более обширного набора прочтений, увеличилось на 12 и 24% (21 456 и 17 342) для стеков на основе ссылок и стеков de novo соответственно.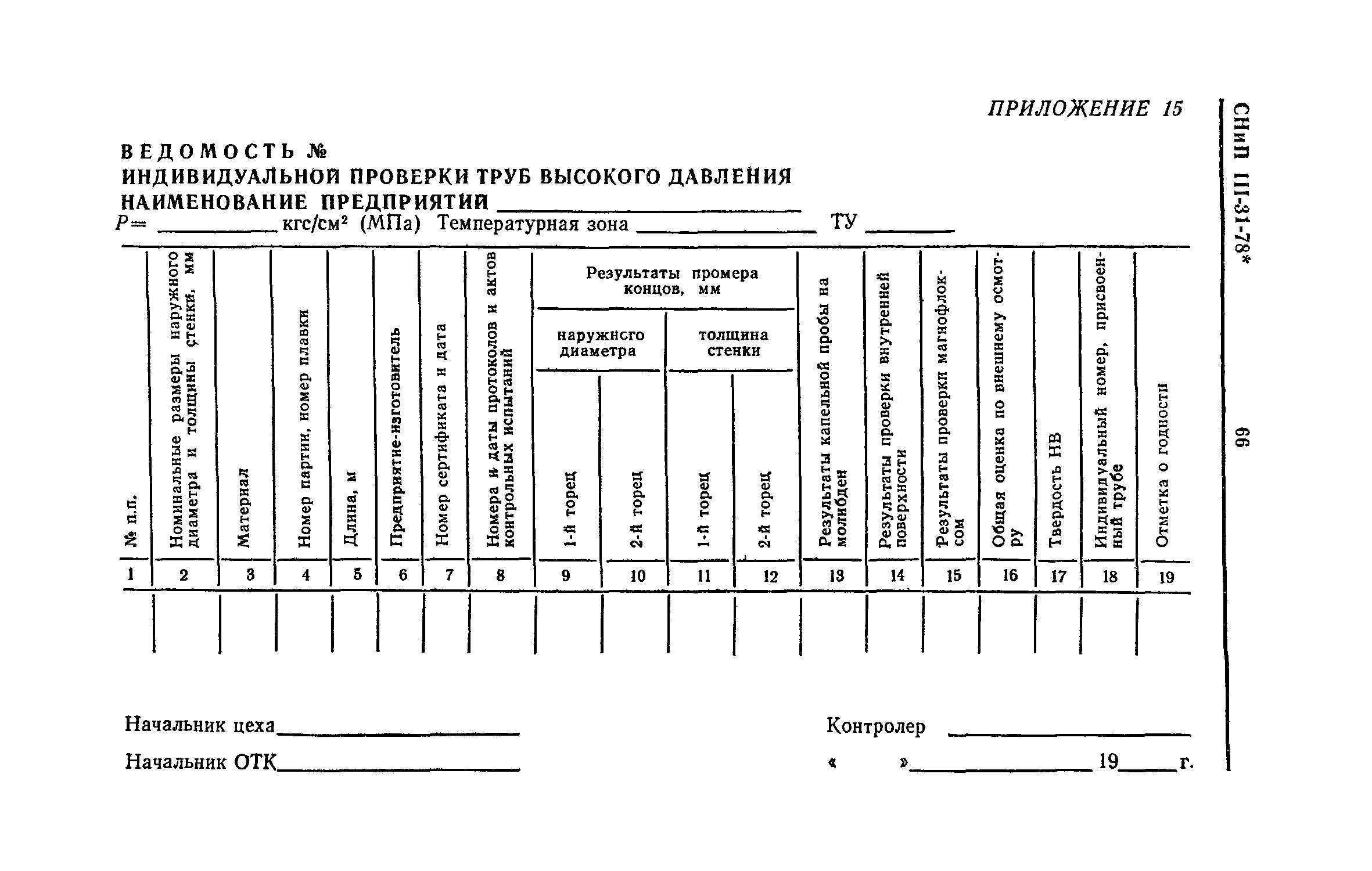 Мы пришли к выводу, что низкая производительность инструмента демультиплексирования Stacks является важной причиной уменьшения количества SNP, вызываемых Stacks.
Мы пришли к выводу, что низкая производительность инструмента демультиплексирования Stacks является важной причиной уменьшения количества SNP, вызываемых Stacks.
GBS с использованием различных платформ секвенирования
Чтобы сравнить вызов SNP с использованием различных технологий секвенирования, мы провели GBS на тех же 24 образцах сои на платформе Ion Torrent. В отличие от ридов Illumina, которые имеют одинаковую длину (100 п.н.), риды Ion Torrent имеют длину от 50 до 135 п.н. В этом анализе мы использовали только два эталонных конвейера, которые показали лучшие результаты в описанных выше тестах (Fast-GBS и TASSEL-GBS v2) с использованием 38 миллионов чтений Ion Torrent.Как видно из Таблицы 5, количество SNP, вызванных в каждом конвейере с одинаковыми уровнями допустимости отсутствующих данных (≤80%) и частотой минорных аллелей (≥0,05), было очень схожим (~ 23 тыс. в обоих случаях). Как и выше, Fast-GBS назвал большее количество вариантов, так как он назвал в общей сложности более 2000 вставок в дополнение к SNP. По времени вычислений Fast-GBS был более чем в два раза быстрее, чем TASSEL-GBS v2 (1 ч 41 мин против 3 ч 39 мин), при этом он использовал на 15 % больше дискового пространства (20 Гб против 17 Гб).
По времени вычислений Fast-GBS был более чем в два раза быстрее, чем TASSEL-GBS v2 (1 ч 41 мин против 3 ч 39 мин), при этом он использовал на 15 % больше дискового пространства (20 Гб против 17 Гб).
Во втором анализе мы измерили количество отсутствующих данных и оценили точность генотипов как путем сравнения генотипов, называемых GBS, с генотипами, полученными в результате повторного секвенирования, так и путем оценки степени гетерозиготности в этих линиях, которые предположительно гомозиготны.Как видно из таблицы 6, доля отсутствующих данных была относительно одинаковой для двух конвейеров (37% против 33%). В этом анализе TASSEL-GBS v2 вызвал больше гетерозиготных генотипов, чем Fast-GBS (6,6% против 4,5%). Также TASSEL-GBS v2 назвал гораздо больше локусов с большей долей (> 50%) гетерозиготных генотипов, чем Fast-GBS (4831 против 861). В этом анализе Fast-GBS снова добился наивысшей точности определения генотипов (95,2%) по сравнению с 91,1% при использовании TASSEL-GBS v2.
Наконец, мы сравнили перекрытие каталогов SNP, полученных с использованием двух платформ секвенирования (Illumina и Ion Torrent).Как показано на рис. 3, при использовании Fast-GBS мы обнаружили, что 69% (16 416 из 23 792 SNP) SNP, полученных из ридов Ion Torrent, также присутствовали в каталоге SNP, полученных с помощью ридов Illumina. И наоборот, из всех SNP, вызванных с помощью чтения Illumina (34 953 SNP), 47% были общими с каталогом Ion Torrent. Используя TASSEL-GBS v2, немного меньшая доля (54%) (12 377 из 22 921 SNP) SNP, вызванных из ридов Ion Torrent, также была получена с использованием ридов Illumina. И наоборот, аналогичная доля (44%) SNP, вызванных с использованием ридов Illumina, была общей с теми, которые были вызваны с помощью ридов Ion Torrent.Мы обнаружили, что использование чтения Ion Torrent приводит к большему количеству неточных SNP по сравнению с чтением Illumina. При использовании чтения Illumina только 23,7% и 12,9% неточных SNP, вызванных TASSEL-GBS v2 и Fast-GBS, имели уникальную позицию, в то время как при использовании чтения Ion Torrent эта доля увеличилась до 76% и 87% для TASSEL-GBS v2 и Fast-GBS. ГБС соответственно. С другой стороны, количество неточных SNP из-за паралогии и повторяющихся областей было одинаковым для обеих технологий. Основываясь на этих результатах, мы делаем вывод, что наблюдаемое увеличение количества неточных SNP с уникальным положением (не из-за какой-либо повторяющейся последовательности) связано с более высокой частотой ошибок секвенирования в чтениях Ion Torrent.
ГБС соответственно. С другой стороны, количество неточных SNP из-за паралогии и повторяющихся областей было одинаковым для обеих технологий. Основываясь на этих результатах, мы делаем вывод, что наблюдаемое увеличение количества неточных SNP с уникальным положением (не из-за какой-либо повторяющейся последовательности) связано с более высокой частотой ошибок секвенирования в чтениях Ion Torrent.
Рис. 3.
Диаграмма Венна для перекрытия SNP, вызванных с использованием двух разных конвейеров биоинформатики (a) Перекрытие SNP, вызванных с помощью Fast-GBS с использованием чтения Illumina и Ion Torrent. (b) Перекрытие SNP, вызванных с помощью TASSEL-GBS v2 с использованием чтения Illumina и Ion Torrent. Проценты указывают предполагаемую точность для всех групп SNP (уникальных или общих).
https://doi.org/10.1371/journal.pone.0161333.g003
В заключение, степень перекрытия между платформами секвенирования была одинаковой при использовании обоих конвейеров, но намного ниже, чем перекрытие между конвейерами, использующими одну и ту же платформу секвенирования.
Обсуждение
Гибкость и низкая стоимость методов генотипирования, основанных на NGS, делают эти инструменты превосходными для многих приложений и исследовательских вопросов в области генетики, селекции и биоразнообразия [3, 6, 23–25]. В настоящее время GBS, по-видимому, предпочтительнее в сельскохозяйственных науках (селекция растений и животных), тогда как RAD-Seq, по-видимому, является более распространенным подходом в области экологии [1]. Какой бы подход к подготовке библиотеки ни был выбран для снижения сложности перед секвенированием, необходимо использовать биоинформатику для извлечения полезной информации о локусах SNP и генотипах из огромного количества ридов коротких последовательностей [1, 26].Именно на этом этапе выбор аналитического метода будет иметь наибольшее влияние на количество и качество получаемой генотипической информации. К сожалению, на сегодняшний день в нескольких исследованиях систематически сравнивались конвейеры вызова SNP для GBS и сравнивалась их эффективность, точность и степень перекрытия.
Первый вопрос, который возникает, касается использования de novo по сравнению с эталонными методами. В отсутствие эталонного генома нет другого выбора, кроме как использовать один из двух широко распространенных в настоящее время инструментов: UNEAK и Stacks.Хотя для этого используются разные алгоритмы, эти два конвейера концептуально схожи в том, что они стремятся сначала установить каталоги идентичных прочтений, а затем искать тесно связанные прочтения, которые потенциально являются аллелями в одном и том же локусе. В условиях, используемых в этой работе, UNEAK значительно превзошел Stacks в том, что он генерировал на 82% больше SNP (~ 25 тыс. против ~ 13 тыс.). С качественной точки зрения оба пайплайна de novo показали себя одинаково хорошо с точки зрения отсутствующих данных (~40%) и генотипической точности (~94%).Это сопоставимо с результатами, полученными Lu et al. (2013) у кукурузы, где было подсчитано, что 92% вызовов генотипа были точными и что эта доля может быть увеличена до 96,2% путем фильтрации SNP с MAF > 0,3 в сегрегирующей биродительской популяции [16]. Оба конвейера de novo могут работать довольно быстро и относительно консервативны в своих вызовах SNP, что приводит к набору данных высокого качества. Таким образом, для подавляющего большинства видов, для которых нет эталонного генома в настоящее время или в обозримом будущем, инструменты вызова SNP de novo работают очень хорошо с точки зрения точности, но UNEAK даст почти в два раза больше SNP.
Оба конвейера de novo могут работать довольно быстро и относительно консервативны в своих вызовах SNP, что приводит к набору данных высокого качества. Таким образом, для подавляющего большинства видов, для которых нет эталонного генома в настоящее время или в обозримом будущем, инструменты вызова SNP de novo работают очень хорошо с точки зрения точности, но UNEAK даст почти в два раза больше SNP.
Однако картина производительности трубопроводов de novo в этом сравнении может быть слишком радужной. Действительно, ради единообразия мы использовали одни и те же параметры фильтрации (MinMAF≥0,05, MaxMD = 80% и minDP≥2) как для de novo , так и для эталонных конвейеров. Но такая высокая устойчивость к отсутствующим данным может оказаться нереалистичной в случае конвейеров de novo . Ранее мы показали, что импутация отсутствующих данных очень эффективна и точна для плотного набора SNP, полученного с использованием эталонного конвейера [18].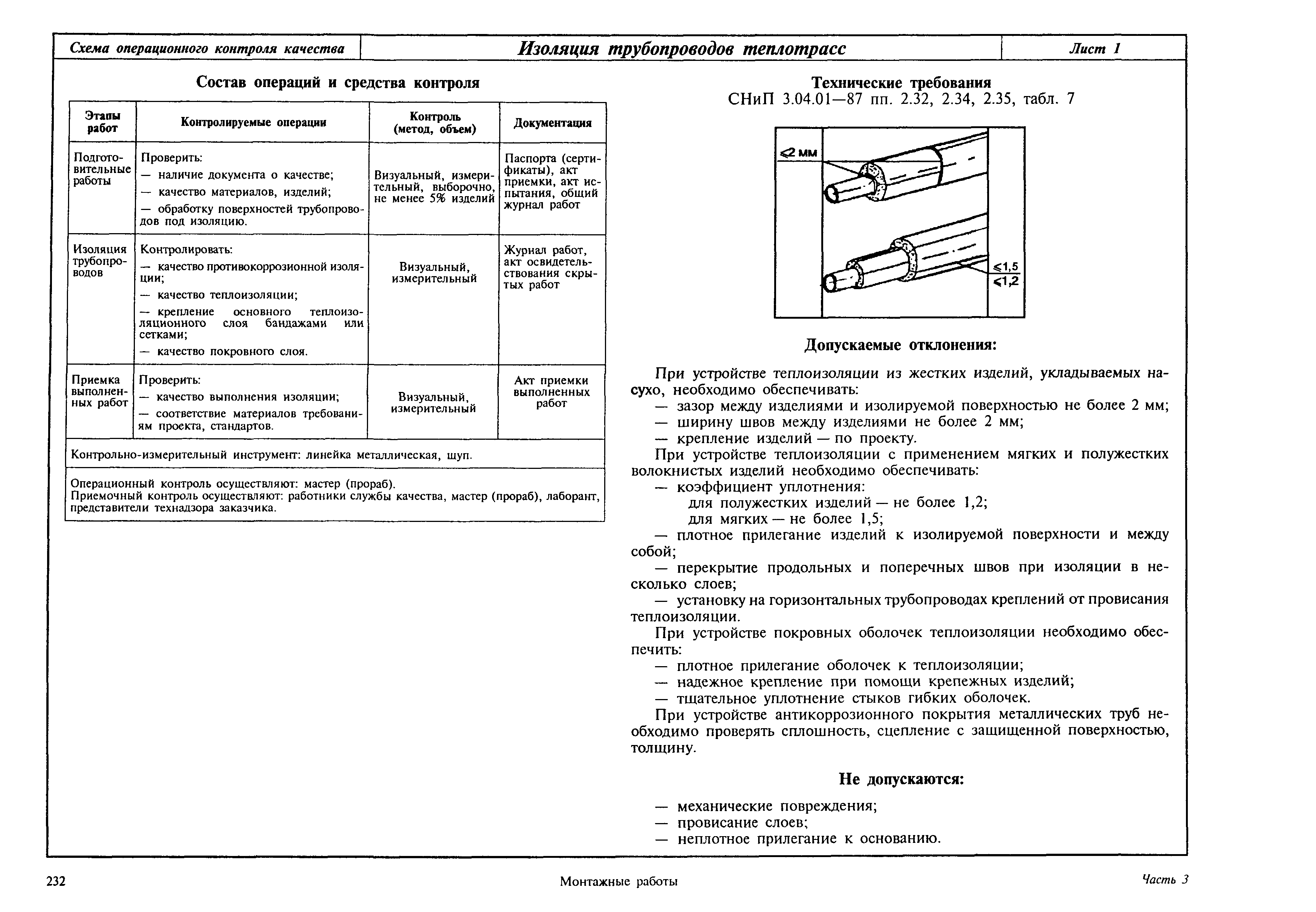 В случае конвейеров de novo при отсутствии информации о местоположении различных SNP и структуре гаплотипов вменение является гораздо более сложной задачей. По этой причине большинство пользователей конвейеров de novo устанавливают более низкий потолок для максимального объема недостающих данных, обычно между 20% и не более 50% [16, 19, 27]. При использовании данных последовательности GBS, используемых в этой работе, допуск до 20% отсутствующих данных существенно снижает количество SNP, которые можно вызвать с использованием обоих конвейеров de novo (~ 5 тыс. SNP; данные не показаны).В этих более реалистичных условиях (с учетом необходимого вменения отсутствующих данных) мы обнаружили, что эталонные пайплайны дали примерно в 5–7 раз больше высококачественных маркеров SNP (~ 5 тыс. маркеров против 25 тыс.–35 тыс.).
В случае конвейеров de novo при отсутствии информации о местоположении различных SNP и структуре гаплотипов вменение является гораздо более сложной задачей. По этой причине большинство пользователей конвейеров de novo устанавливают более низкий потолок для максимального объема недостающих данных, обычно между 20% и не более 50% [16, 19, 27]. При использовании данных последовательности GBS, используемых в этой работе, допуск до 20% отсутствующих данных существенно снижает количество SNP, которые можно вызвать с использованием обоих конвейеров de novo (~ 5 тыс. SNP; данные не показаны).В этих более реалистичных условиях (с учетом необходимого вменения отсутствующих данных) мы обнаружили, что эталонные пайплайны дали примерно в 5–7 раз больше высококачественных маркеров SNP (~ 5 тыс. маркеров против 25 тыс.–35 тыс.).
Учитывая растущую доступность эталонных геномов экономически важных сельскохозяйственных культур и животных, нам необходимо задаться вопросом, какой из доступных эталонных конвейеров дает лучший каталог SNP как с точки зрения обилия маркеров, так и с точки зрения их точности. Среди пяти конвейеров, основанных на эталонах, Fast-GBS можно запустить быстро, что привело к самой высокой точности генотипирования для очень большого количества локусов SNP (около 35 000) в дополнение к почти 4000 инделей.Исходя из этих соображений, он кажется предпочтительным, по крайней мере, в случае сои и, вероятно, также для других видов с аналогичными геномными и репродуктивными характеристиками.
Среди пяти конвейеров, основанных на эталонах, Fast-GBS можно запустить быстро, что привело к самой высокой точности генотипирования для очень большого количества локусов SNP (около 35 000) в дополнение к почти 4000 инделей.Исходя из этих соображений, он кажется предпочтительным, по крайней мере, в случае сои и, вероятно, также для других видов с аналогичными геномными и репродуктивными характеристиками.
Из протестированных пайплайнов TASSEL-GBSv1 отличался от остальной группы по количеству вызываемых локусов SNP (на 50–100 % больше, чем у других), но это происходило за счет точности, поскольку это был единственный трубопровод, чьи генотипические вызовы были точными менее чем в 90% случаев (76,1%). Поскольку отличить истинный генотип от ложного непросто, мы утверждаем, что TASSEL-GBSv1 недостаточно точен, чтобы его можно было использовать отдельно.В предыдущей работе большой результирующий каталог SNP часто «фильтровался» путем отбрасывания маркеров, которые не вели себя должным образом в сегрегирующей популяции [6]. Это, по-видимому, помогло отбросить «ложные» маркеры, возникшие в результате смешения аллелей (в одном локусе) и прочтений, полученных из паралогичных локусов. Мы предположили, что основной причиной снижения точности является тот факт, что TASSEL-GBSv1 обрезает все чтения до одинаковой длины в 64 базы, создавая таким образом короткие теги, которые подвергаются повышенному риску сопоставления с несколькими или ошибочными местоположениями.Конвейеры, использующие более длительные чтения, не проявляли этой проблемы и обычно имели как минимум в 10 раз меньше операций чтения, сопоставленных с несколькими местоположениями. Например, несмотря на то, что у TASSEL-GBS v1 много общего, когда TASSEL-GBS v2 запускали в условиях, позволяющих использовать более длинные теги (в нашем случае 92 основания), надежность генотипов значительно возросла.
Это, по-видимому, помогло отбросить «ложные» маркеры, возникшие в результате смешения аллелей (в одном локусе) и прочтений, полученных из паралогичных локусов. Мы предположили, что основной причиной снижения точности является тот факт, что TASSEL-GBSv1 обрезает все чтения до одинаковой длины в 64 базы, создавая таким образом короткие теги, которые подвергаются повышенному риску сопоставления с несколькими или ошибочными местоположениями.Конвейеры, использующие более длительные чтения, не проявляли этой проблемы и обычно имели как минимум в 10 раз меньше операций чтения, сопоставленных с несколькими местоположениями. Например, несмотря на то, что у TASSEL-GBS v1 много общего, когда TASSEL-GBS v2 запускали в условиях, позволяющих использовать более длинные теги (в нашем случае 92 основания), надежность генотипов значительно возросла.
Эталонная версия Stacks — это еще один конвейер, который выделяется тем, что вызывает гораздо меньше SNP, чем другие. При исследовании различных шагов, необходимых для перехода от последовательностей к SNP, мы обнаружили, что стеки потеряли ~ 20% чтений на этапе демультиплексирования, т. е.е. некоторые считывания штрих-кода не относились к образцу и просто отбрасывались на последующих этапах. Это, очевидно, привело к сопутствующему уменьшению количества названных SNP (~ 19 тыс. против ~ 25 тыс.). Об этой плохой производительности шага демультиплексирования Stacks ранее сообщалось Хертеном и др. [28].
е.е. некоторые считывания штрих-кода не относились к образцу и просто отбрасывались на последующих этапах. Это, очевидно, привело к сопутствующему уменьшению количества названных SNP (~ 19 тыс. против ~ 25 тыс.). Об этой плохой производительности шага демультиплексирования Stacks ранее сообщалось Хертеном и др. [28].
По нашему мнению, полногеномное измерение точности наборов данных GBS, полученных из различных каналов биоинформатики, представляет собой важный и ключевой вклад в эту работу. Его оценивали путем прямого сравнения с данными ресеквенирования всего генома.Во многих предыдущих исследованиях оценка генотипической точности часто достигалась косвенным измерением [16] или выполнялась на очень небольшом подмножестве локусов SNP [9]. Как правило, сообщается об уровнях точности генотипа в диапазоне от 92 до 98% с небольшими различиями, наблюдаемыми между видами и типами популяций [9, 16, 19]. Преимущество использования данных повторного секвенирования таким образом заключается в том, что мы можем напрямую оценить точность данных GBS, полученных из разных конвейеров.
Еще одно важное соображение заключается в том, согласуются ли каталоги SNP, созданные с использованием разных конвейеров и разных технологий секвенирования.При использовании одной технологии секвенирования (Illumina) мы обнаружили, что около 80% или более SNP, вызываемых большинством конвейеров, также присутствовали в каталоге SNP, полученном из Fast-GBS. Таким образом, эти конвейеры в значительной степени согласуются с локусами, полиморфными в пределах данного набора зародышевой плазмы. Единственным исключением был TASSEL-GBS v1, так как только четверть SNP, представленных в результирующем каталоге, также присутствовала в наборе, полученном с помощью Fast-GBS. Вероятно, это связано с более короткими последовательностями (всего 64 п.н.) и большим количеством «ложных» SNP, поскольку этот конвейер оказался наименее точным из всех.При использовании одного и того же конвейера для анализа данных, полученных с помощью двух технологий секвенирования (Illumina и Ion Torrent), мы обычно обнаруживали, что перекрытие между каталогами SNP варьировалось примерно от 50 до 70%. Таким образом, выбор используемой технологии секвенирования привел к большей изменчивости в каталоге полученных SNP, чем выбор конвейера, используемого для одного набора прочтений. На первый взгляд может показаться, что это противоречит выводам, сделанным Mascher et al. (2013), которые обнаружили, что каталоги SNP, созданные с использованием двух конвейеров (TASSEL-GBS v1 и SAMtools), отличаются больше, чем каталоги, полученные с использованием разных технологий секвенирования (Illumina и Ion Torrent) [19].На наш взгляд, это скорее отражение ограничений TASSEL-GBS v1 (из-за коротких тегов). Когда мы рассматриваем более широкий набор эталонных пайплайнов, они, как правило, обеспечивают очень хорошее перекрытие в непокрытых локусах SNP.
Таким образом, выбор используемой технологии секвенирования привел к большей изменчивости в каталоге полученных SNP, чем выбор конвейера, используемого для одного набора прочтений. На первый взгляд может показаться, что это противоречит выводам, сделанным Mascher et al. (2013), которые обнаружили, что каталоги SNP, созданные с использованием двух конвейеров (TASSEL-GBS v1 и SAMtools), отличаются больше, чем каталоги, полученные с использованием разных технологий секвенирования (Illumina и Ion Torrent) [19].На наш взгляд, это скорее отражение ограничений TASSEL-GBS v1 (из-за коротких тегов). Когда мы рассматриваем более широкий набор эталонных пайплайнов, они, как правило, обеспечивают очень хорошее перекрытие в непокрытых локусах SNP.
Выводы, сделанные в этой работе, вероятно, распространяются на другие организмы, имеющие сходные геномные особенности (геном среднего размера, диплоидный). Можно ожидать, что виды, пережившие недавние события дупликации всего генома, будут представлять большую проблему, поскольку в таких случаях, вероятно, возрастет риск смешения аллелей в одном и том же локусе и паралогах.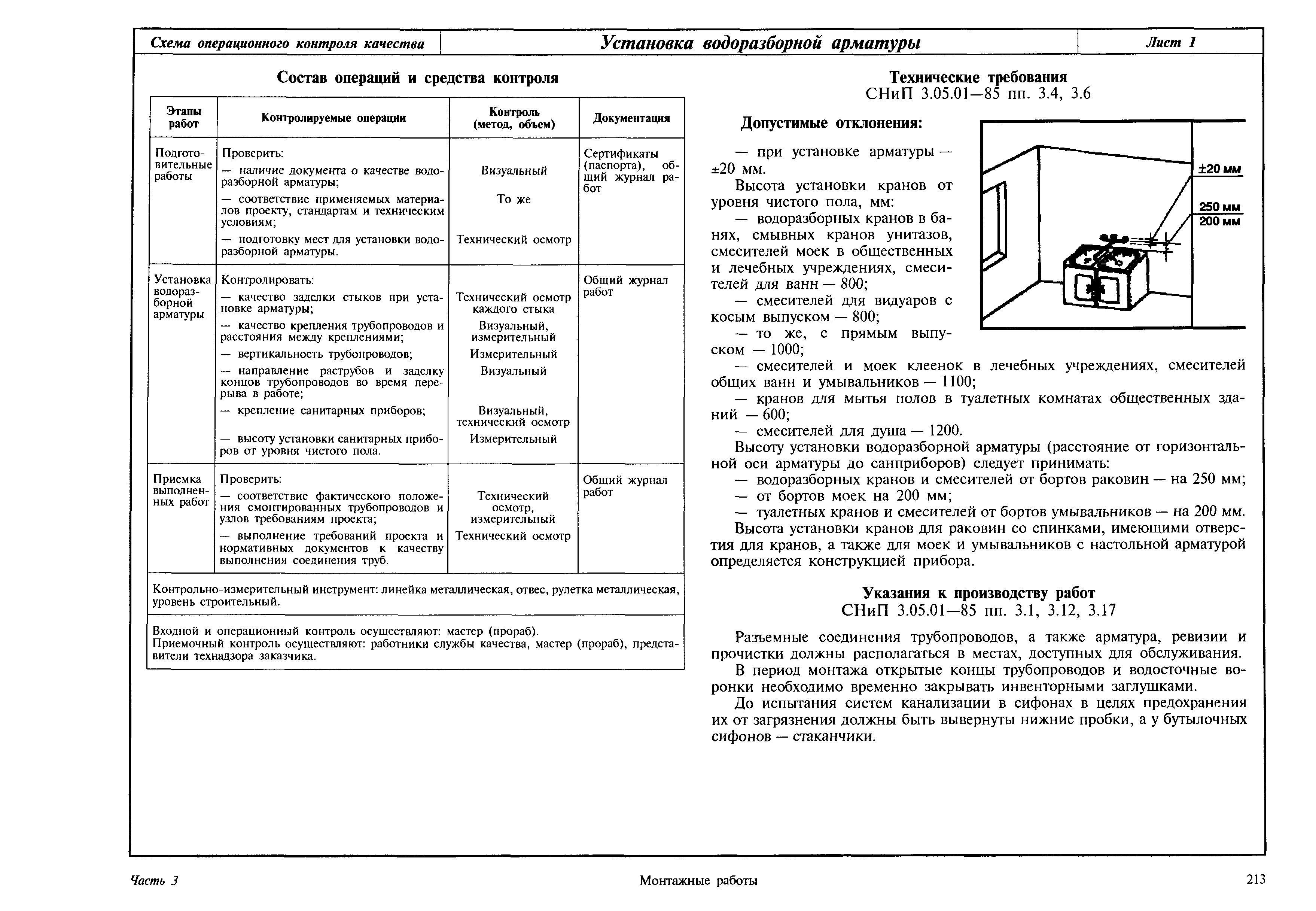 У видов, у которых такие события происходили в более отдаленном прошлом, у паралогов было больше возможностей расходиться, что облегчало правильное картирование прочтений.
У видов, у которых такие события происходили в более отдаленном прошлом, у паралогов было больше возможностей расходиться, что облегчало правильное картирование прочтений.
Таким образом, невозможно разработать единый конвейер, одинаково подходящий для любой ситуации. Именно здесь для пользователей становится важным иметь возможность изменять различные параметры в процессе вызова SNP. К сожалению, не все пайплайны в этом отношении одинаково «прозрачны» и дают одинаковые возможности для изменений.С одной стороны, UNEAK и TASSEL-GBS предлагают очень хорошую производительность, но полагаются на некоторые специально созданные инструменты или алгоритмы, которые пользователь не может легко изменить (например, для демультиплексирования и вариантного вызова). Кроме того, промежуточные файлы данных не всегда легкодоступны, что затрудняет исследование конкретных проблем. С другой стороны, IGST и Fast-GBS объединяют набор существующих инструментов, для которых пользователь может изменять параметры/параметры по своему желанию, а промежуточные файлы легко доступны.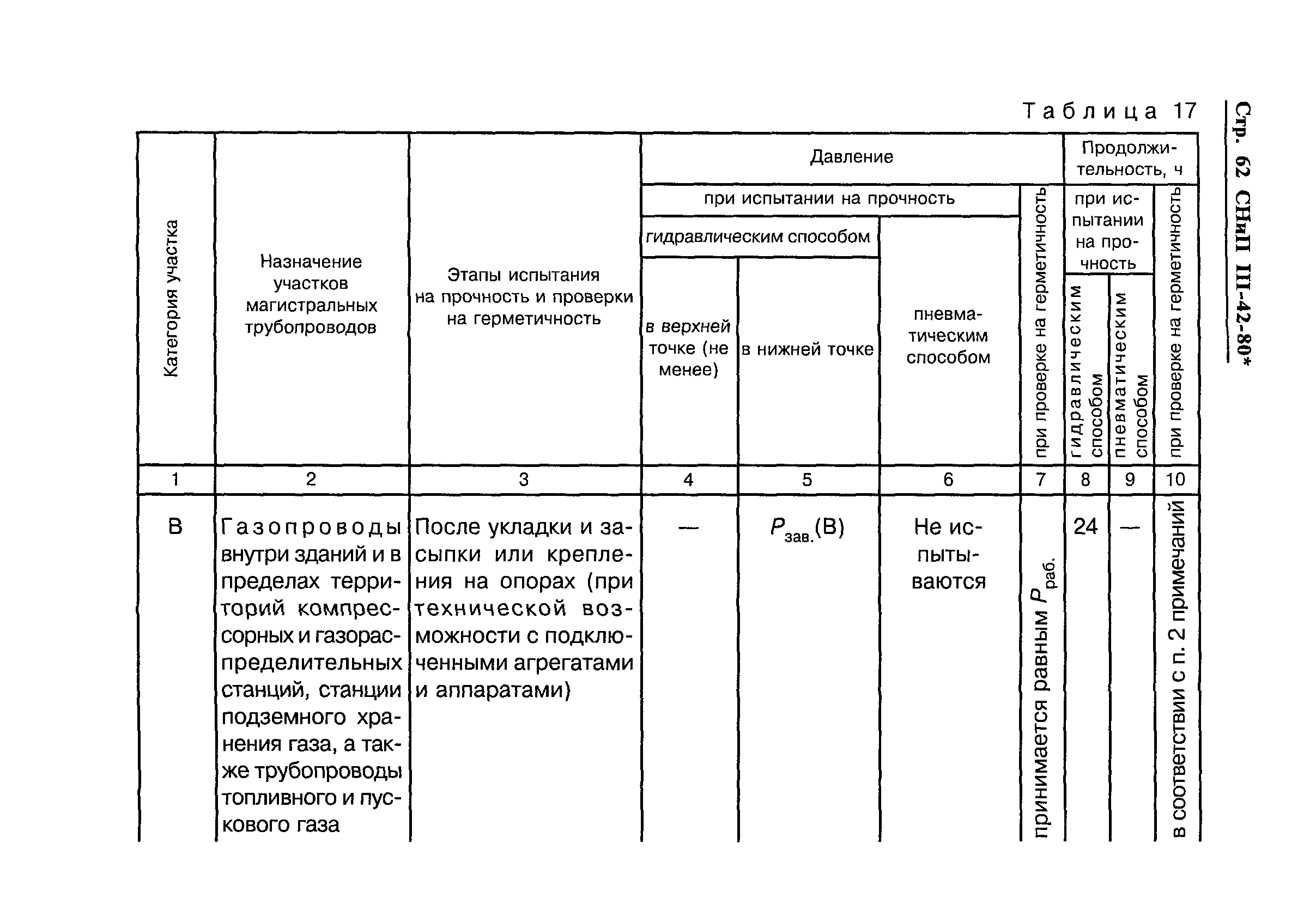 В этом спектре, на наш взгляд, Stacks предлагает промежуточный уровень прозрачности.
В этом спектре, на наш взгляд, Stacks предлагает промежуточный уровень прозрачности.
Наконец, хотя быстро приближается полногеномное секвенирование целых популяций, мы полагаем, что описанные здесь методы, вероятно, останутся бесценными в ближайшие годы в популяционной геномике, селекции, картировании и сборке эталонных последовательностей генома, особенно для немодельных организмы.
Вклад авторов
- Концептуализация: DT FB.
- Формальный анализ: DT.
- Получение финансирования: FB.
- Расследование: DT.
- Надзор: ФБ.
- Письмо — первоначальный проект: DT FB.
- Написание — рецензирование и редактирование: DT JL FB.
Каталожные номера
- 1.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM и Blaxter ML. Открытие полногеномных генетических маркеров и генотипирование с использованием секвенирования нового поколения. Природа. 2011 г.;
- 2. Миллер М. Р., Данхэм Дж. П., Аморес А., Креско В. А. и Джонсон Э. А. Быстрая и экономичная идентификация полиморфизма и генотипирование с использованием маркеров ДНК, связанных с сайтом рестрикции (RAD). Геном Res. 17, 240–248 (2007). пмид:17189378
- 3. Бэрд Н.А., Эттер П.Д., Этвуд Т.С. и соавт. (2008)Быстрое открытие SNP и генетическое картирование с использованием секвенированных маркеров RAD. PLoS ONE, 3, e3376. пмид:18852878
- 4. Van Orsouw NJ, Hogers RCJ, Janssen A et al.(2007) Снижение сложности полиморфных последовательностей (CRoPS): новый подход к обнаружению крупномасштабного полиморфизма в сложных геномах. ПЛОС ОДИН, 2, e1172. пмид:18000544
- 5. Андольфатто П. и соавт. Мультиплексное генотипирование дробовика для быстрого и эффективного генетического картирования. Геном Res. 21, 610–617 (2011). пмид:21233398
- 6.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, et al. Надежный и простой подход генотипирования путем секвенирования (GBS) для видов с большим разнообразием.ПЛОС ОДИН. 2011 г.; 6: e19379. пмид:21573248
- 7. Петерсон Б.К., Вебер Дж.Н., Кей Э.Х., Фишер Х.С., Хекстра Х.Е. (2012)Двойной обзор radseq: недорогой метод обнаружения и генотипирования SNP de novo у модельных и немодельных видов. PLoS ONE, 7, e37135. пмид:22675423
- 8. Парчман Т.Л., Гомперт З., Мадж Дж., Шилки Ф.Д., Бенкман К.В., Бюркле К.А. (2012)Полногеномная ассоциативная генетика адаптивного признака скрученной сосны. Молекулярная экология, 21, 2991–3005. пмид:22404645
- 9.Сона Х., Бастьен М., Икира Э., Тардивел А., Легаре Г. и др. Усовершенствованный подход к генотипированию путем секвенирования (GBS), предлагающий повышенную универсальность и эффективность обнаружения SNP и генотипирования. ПЛОС ОДИН 2013; 8(1): e54603. пмид:23372741
- 10.
Кумар С., Бэнкс Т.В. и Клотье С. Открытие SNP с помощью секвенирования нового поколения и его приложений. Международный журнал геномики растений. 2012 г.;
- 11. Ли Х. и Дурбин Р. Быстрое и точное выравнивание коротких считываний с преобразованием Берроуза-Уилера.Биоинформатика. 2009 г.; 25, 1754–1760 гг., середина: 19451168
- 12. Нильсен Р., Пол Дж. С., Альбрехтсен А. и Сонг Ю. С. Генотип и вызов SNP на основе данных секвенирования следующего поколения. Природа Преподобный Жене. 12, 443–451 (2011). пмид:21587300
- 13. Брэдбери П.Дж., Чжан З., Крун Д.Э., Касстевенс Т.М., Рамдосс Ю., Баклер Э.С. TASSEL: программное обеспечение для картирования ассоциаций сложных признаков в различных образцах. Биоинформатика. 2007;23(19):2633–5. пмид:17586829
- 14.Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, et al. (2014) TASSEL-GBS: высокопроизводительное генотипирование с помощью конвейера анализа последовательности. PLoS ONE 9(2): e
. пмид:24587335
- 15.
Catchen J1, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Стеки: набор инструментов для анализа популяционной геномики. Мол Экол. 2013 июнь; 22 (11): 3124–40. Epub 2013 24 мая. pmid:23701397
- 16. Лу Ф., Липка А.Е., Глаубиц Дж., Элшир Р., Черни Дж.Х. и соавт. (2013) Геномное разнообразие, плоидность и эволюция проса проса: новый взгляд на сетевой протокол обнаружения SNP.PLoS Genet 9(1): e1003215. пмид:23349638
- 17. Ротберг Дж. М., Хинц В., Рерик Т. М., Шульц Дж., Милески В. и соавт. 2011. Интегрированное полупроводниковое устройство, позволяющее проводить неоптическое секвенирование генома. Природа 475, 348–352. пмид:21776081
- 18. Torkamaneh D, Belzile F (2015)Сканирование и заполнение: сверхплотное генотипирование SNP, сочетающее генотипирование с помощью секвенирования, массив SNP и данные полногеномного повторного секвенирования. PLoS ONE 10(7): e0131533. пмид:26161900
- 19.Машер М., Ву С., Аманд П.С., Стейн Н., Польша Дж. (2013) Применение генотипирования путем секвенирования на полупроводниковых платформах секвенирования: сравнение упорядочения генетических и эталонных маркеров в ячмене. PLoS ONE 8(10): e76925. пмид:24098570
- 20. Шмутц Дж., Кэннон С.Б., Шлютер Дж., Ма Дж., Митрос Т. и др. Последовательность генома палеополиплоидной сои. Природа. 2010 г.; 463 (7278): 178–183. пмид:20075913
- 21. Ли Х.*, Хэндсейкер Б.*, Высокер А., Феннелл Т., Руан Дж., Гомер Н., Март Г., Абекасис Г., Дурбин Р. и Подгруппа обработки данных проекта 1000 геномов (2009 г.) Формат выравнивания/карты последовательностей (SAM) и SAMtools. Биоинформатика, 25, 2078–9. [PMID: pmid:19505943]
- 22. The Variant Call Format и VCFtools, Петр Данечек, Адам Аутон, Гонсало Абеказис, Корнелис А. Альберс, Эрик Бэнкс, Марк А. ДеПристо, Роберт Хэндсейкер, Гертон Лунтер, Габор Март, Стивен Т. Шерри, Джилин Маквин, Ричард Дурбин и 1000 Группа анализа проекта геномов, биоинформатика, 2011
- 23.Сегал Д., Викрам П., Сансалони С.П., Ортис С., Пьер С.С., Пейн Т. и др. (2015) Изучение и мобилизация биоразнообразия банка генов для улучшения пшеницы. PLoS ONE 10(7): e0132112. пмид:26176697
- 24. Truong HT, Ramos AM, Yalcin F, de Ruiter M, van der Poel HJA, et al. (2012) Генотипирование на основе последовательностей для обнаружения маркеров и оценки кодоминантности в зародышевой плазме и популяциях. PLoS ONE 7(5): e37565. пмид:22662172
- 25. Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Разработка генетических карт высокой плотности для ячменя и пшеницы с использованием нового двухферментного генотипирования путем секвенирования.PLoS ONE 7(2): e32253. пмид:22389690
- 26. McCormack JE, Hird SM, Zellmer AJ, Carstens BC, Brumfield RT (2013) Применение секвенирования нового поколения в филогеографии и филогенетике. Молекулярная филогенетика и эволюция, 66, 526–538. пмид:22197804
- 27.
Ларсон В.А., Сиб Л.В., Эверетт М.В., Уэйплс Р.К., Темплин В.Д., Сиб Дж.Е. Генотипирование путем секвенирования разрешает поверхностную структуру популяции, чтобы обеспечить сохранение чавычи ( Oncorhynchus tshawytscha ). Эволюционные приложения. 2014;7(3):355–369. пмид:24665338
- 28. Herten K, Hestand MS, Vermeesch JR и Van Houdt JKJ (2015) GBSX: набор инструментов для экспериментального дизайна и демультиплексирования генотипирования с помощью экспериментов по секвенированию. Биоинформатика BMC.
 Природа. 2011 г.;
Природа. 2011 г.; Надежный и простой подход генотипирования путем секвенирования (GBS) для видов с большим разнообразием.ПЛОС ОДИН. 2011 г.; 6: e19379. пмид:21573248
Надежный и простой подход генотипирования путем секвенирования (GBS) для видов с большим разнообразием.ПЛОС ОДИН. 2011 г.; 6: e19379. пмид:21573248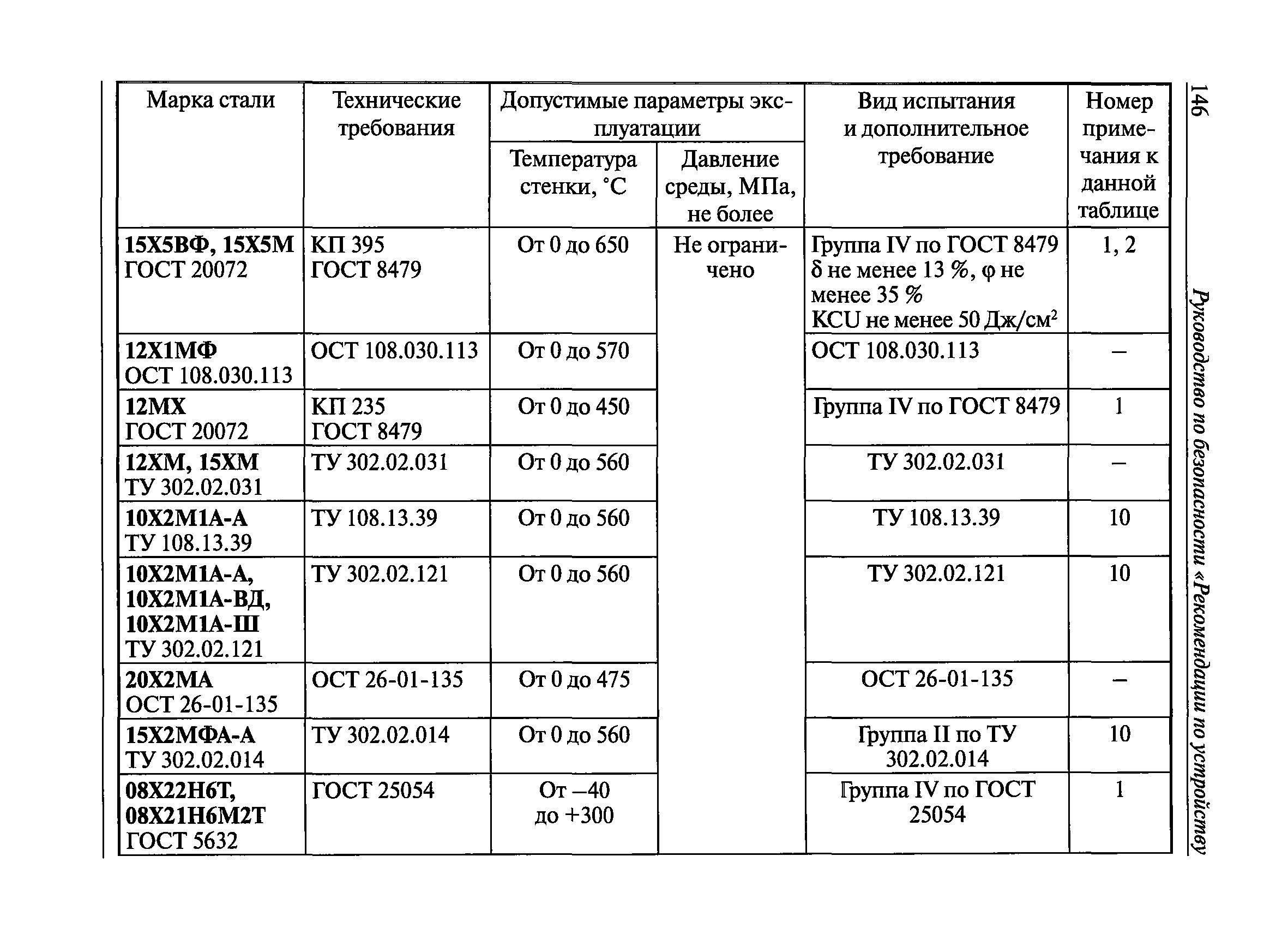 Международный журнал геномики растений. 2012 г.;
Международный журнал геномики растений. 2012 г.; Стеки: набор инструментов для анализа популяционной геномики. Мол Экол. 2013 июнь; 22 (11): 3124–40. Epub 2013 24 мая. pmid:23701397
Стеки: набор инструментов для анализа популяционной геномики. Мол Экол. 2013 июнь; 22 (11): 3124–40. Epub 2013 24 мая. pmid:23701397 PLoS ONE 8(10): e76925. пмид:24098570
PLoS ONE 8(10): e76925. пмид:24098570 PLoS ONE 10(7): e0132112. пмид:26176697
PLoS ONE 10(7): e0132112. пмид:26176697 Эволюционные приложения. 2014;7(3):355–369. пмид:24665338
Эволюционные приложения. 2014;7(3):355–369. пмид:24665338границ | Сравнение конвейеров вызова SNP и платформ NGS для прогнозирования геномных регионов, содержащих гены-кандидаты для образования клубеньков в культивируемом арахисе
Введение
Арахис ( Arachis hypogaea L.) является одной из самых важных масличных культур, выращиваемых во всем мире. Как вид бобовых, арахис может образовывать симбиотические отношения с ризобиями, чтобы биологически фиксировать азот, тем самым уменьшая количество синтетических азотных удобрений, применяемых в вегетационный период. Симбиотический процесс и молекулярные механизмы были тщательно изучены на двух модельных видах бобовых Lotus japonicas и Medicago truncatula , где ризобии проникают в растение-хозяин через внутриклеточный путь корневых волосков (Oldroyd, 2013). Многие гены были охарактеризованы в симбиотическом пути, и также сообщалось, что некоторые фрагменты малых РНК ризобий играют регулирующую роль (Ren et al., 2019). У арахиса ризобии заражают растения через межклеточную щель, которая менее изучена и недостаточно изучена (Peng et al., 2017a). Неклубеньковые (Nod-) растения арахиса, о которых впервые сообщили Горбет и Бертон (1979), являются важным материалом для анализа генетических факторов образования клубеньков у арахиса. Растения Nod-peanut были впервые идентифицированы в популяции F 3 в результате скрещивания двух клубеньковых (Nod+) генотипов 487A-4-1-2 и PI 262090 (Gorbet and Burton, 1979).Несколько моделей наследования генов были впоследствии предложены путем изучения коэффициентов сегрегации в популяциях, сегрегирующих по нодуляции, включая двухгенную (Nigam et al., 1980) и трехгенную (Dutta and Reddy, 1988; Gallo-Meagher et al., 2001). ) модели. Однако гены нодуляции не были ни идентифицированы, ни охарактеризованы.
Многие гены были охарактеризованы в симбиотическом пути, и также сообщалось, что некоторые фрагменты малых РНК ризобий играют регулирующую роль (Ren et al., 2019). У арахиса ризобии заражают растения через межклеточную щель, которая менее изучена и недостаточно изучена (Peng et al., 2017a). Неклубеньковые (Nod-) растения арахиса, о которых впервые сообщили Горбет и Бертон (1979), являются важным материалом для анализа генетических факторов образования клубеньков у арахиса. Растения Nod-peanut были впервые идентифицированы в популяции F 3 в результате скрещивания двух клубеньковых (Nod+) генотипов 487A-4-1-2 и PI 262090 (Gorbet and Burton, 1979).Несколько моделей наследования генов были впоследствии предложены путем изучения коэффициентов сегрегации в популяциях, сегрегирующих по нодуляции, включая двухгенную (Nigam et al., 1980) и трехгенную (Dutta and Reddy, 1988; Gallo-Meagher et al., 2001). ) модели. Однако гены нодуляции не были ни идентифицированы, ни охарактеризованы. Исследование транскриптома с использованием образцов корней из двух наборов рекомбинантных инбредных линий (RIL) с фенотипом Nod+ и Nod- выявило сотни дифференциально экспрессируемых генов (DEG) при заражении ризобиями (Peng et al., 2017а). Кроме того, те же материалы были морфологически и генетически охарактеризованы, чтобы начать исследования генов клубеньков арахиса (Peng et al., 2018). Всего для генетической характеристики было использовано 188 маркеров простых повторов последовательностей (SSR), и только несколько полиморфных SSR были получены между RIL из-за их высокого генетического сходства. Впоследствии были построены графические карты генотипов RIL, показывающие геномные области-кандидаты, контролирующие образование клубеньков арахиса, и в общей сложности между двумя наборами RIL было выявлено 22 хромосомных участка, потенциально связанных с образованием клубеньков.Однако при ограниченном количестве маркеров карты имели низкое разрешение, что затрудняло дальнейшую точную картографию.
Исследование транскриптома с использованием образцов корней из двух наборов рекомбинантных инбредных линий (RIL) с фенотипом Nod+ и Nod- выявило сотни дифференциально экспрессируемых генов (DEG) при заражении ризобиями (Peng et al., 2017а). Кроме того, те же материалы были морфологически и генетически охарактеризованы, чтобы начать исследования генов клубеньков арахиса (Peng et al., 2018). Всего для генетической характеристики было использовано 188 маркеров простых повторов последовательностей (SSR), и только несколько полиморфных SSR были получены между RIL из-за их высокого генетического сходства. Впоследствии были построены графические карты генотипов RIL, показывающие геномные области-кандидаты, контролирующие образование клубеньков арахиса, и в общей сложности между двумя наборами RIL было выявлено 22 хромосомных участка, потенциально связанных с образованием клубеньков.Однако при ограниченном количестве маркеров карты имели низкое разрешение, что затрудняло дальнейшую точную картографию. С помощью технологий секвенирования следующего поколения (NGS) плотность карты может быть дополнительно улучшена.
С помощью технологий секвенирования следующего поколения (NGS) плотность карты может быть дополнительно улучшена.
Peanut представляет собой аллотетраплоид (2 n = 2x = 40; AABB; ~2,7 Гб) с двумя субгеномами, A и B, полученными из A. duranensis и A. ipaensis соответственно (Bertioli et al. ., 2016). Доступные эталонные геномы двух диплоидных предков сделали полногеномное повторное секвенирование (WGRS) применимым подходом для высокопроизводительного генотипирования, которое использовалось для генотипирования популяции с двумя родителями для построения генетической карты высокой плотности и идентификации генов-кандидатов устойчивости к болезням в арахис (Агарвал и др., 2018). Каждый образец был секвенирован с 2-5-кратным покрытием. Однако, учитывая большой размер генома и высокое содержание повторяющихся последовательностей в геноме арахиса, WGRS все еще может быть не самой рентабельной стратегией для обнаружения генетических вариаций, поскольку стоимость образца по-прежнему высока, особенно если ожидается высокий охват (Schwarze). и др., 2018). В качестве альтернативы, другие методы генотипирования с поддержкой NGS с уменьшенной сложностью генома могут быть экономически эффективными для высокопроизводительного генотипирования, например секвенирование РНК (RNA-seq) (Clevenger et al., 2015; Chopra et al., 2016), генотипирование путем секвенирования (GBS) (Tseng et al., 2016) и секвенирование с обогащением мишеней (TES) (Peng et al., 2017b), которые обнаруживают генетические вариации у репрезентативной части геном. Кроме того, массив Axiom Arachis2 с 47 837 SNP может быть экономичным и простым методом высокопроизводительного генотипирования (Clevenger et al., 2018), хотя он ограничен только известными однонуклеотидными полиморфизмами (SNP).
и др., 2018). В качестве альтернативы, другие методы генотипирования с поддержкой NGS с уменьшенной сложностью генома могут быть экономически эффективными для высокопроизводительного генотипирования, например секвенирование РНК (RNA-seq) (Clevenger et al., 2015; Chopra et al., 2016), генотипирование путем секвенирования (GBS) (Tseng et al., 2016) и секвенирование с обогащением мишеней (TES) (Peng et al., 2017b), которые обнаруживают генетические вариации у репрезентативной части геном. Кроме того, массив Axiom Arachis2 с 47 837 SNP может быть экономичным и простым методом высокопроизводительного генотипирования (Clevenger et al., 2018), хотя он ограничен только известными однонуклеотидными полиморфизмами (SNP).
Поскольку геномы A и B арахиса очень похожи со средней идентичностью 93.11% (Bertioli et al., 2016), было большой проблемой идентифицировать аллельные SNP из-за смешивающего эффекта гомеологичных SNP между двумя субгеномами (Clevenger et al., 2015). Для решения этой проблемы было разработано несколько стратегий и инструментов. Одним из вариантов уменьшения количества гомеологичных SNP является использование исключительно уникально картированных прочтений для последующего вызова SNP (Zhou et al., 2014; Peng et al., 2017b), что привело к уменьшению количества идентифицированных полезных SNP. В качестве альтернативы было разработано несколько других методов, которые могут использовать общие сопоставленные чтения для вызова SNP и впоследствии отфильтровывать гомеологичные SNP.Например, SWEEP (Clevenger and Ozias-Akins, 2015), который использует гомеологичные SNP в качестве якоря для дифференциации аллельных SNP, был успешно применен к арахису (Clevenger et al., 2017; Pandey et al., 2017) с валидацией. уровень 85% с помощью секвенирования по Сэнгеру и более 95% с помощью данных моделирования (Clevenger and Ozias-Akins, 2015). Кроме того, был разработан инструмент машинного обучения под названием SNP-ML для прогнозирования аллельных SNP со степенью достоверности 75–98% (Korani et al., 2019). Была разработана улучшенная версия SWEEP, названная HAPLOSWEEP, которая применяет основанный на гаплотипах метод для выявления аллельных полиморфизмов между генотипами (Clevenger et al.
Одним из вариантов уменьшения количества гомеологичных SNP является использование исключительно уникально картированных прочтений для последующего вызова SNP (Zhou et al., 2014; Peng et al., 2017b), что привело к уменьшению количества идентифицированных полезных SNP. В качестве альтернативы было разработано несколько других методов, которые могут использовать общие сопоставленные чтения для вызова SNP и впоследствии отфильтровывать гомеологичные SNP.Например, SWEEP (Clevenger and Ozias-Akins, 2015), который использует гомеологичные SNP в качестве якоря для дифференциации аллельных SNP, был успешно применен к арахису (Clevenger et al., 2017; Pandey et al., 2017) с валидацией. уровень 85% с помощью секвенирования по Сэнгеру и более 95% с помощью данных моделирования (Clevenger and Ozias-Akins, 2015). Кроме того, был разработан инструмент машинного обучения под названием SNP-ML для прогнозирования аллельных SNP со степенью достоверности 75–98% (Korani et al., 2019). Была разработана улучшенная версия SWEEP, названная HAPLOSWEEP, которая применяет основанный на гаплотипах метод для выявления аллельных полиморфизмов между генотипами (Clevenger et al. , 2018), а уровень проверки составил 74 % благодаря генотипированию с помощью массива Axiom Arachis2 . С этими методами и инструментами, доступными для сообщества арахиса, в настоящее время не проводилось исследований для сравнения этих методов вызова и фильтрации SNP или для сравнения эффектов картирования считываний с конкатенированным геномом A + B или с геномами A и B по отдельности (A /Б).
, 2018), а уровень проверки составил 74 % благодаря генотипированию с помощью массива Axiom Arachis2 . С этими методами и инструментами, доступными для сообщества арахиса, в настоящее время не проводилось исследований для сравнения этих методов вызова и фильтрации SNP или для сравнения эффектов картирования считываний с конкатенированным геномом A + B или с геномами A и B по отдельности (A /Б).
В этом исследовании для изучения генетических факторов и генетических областей, контролирующих образование клубеньков у арахиса, SNP были идентифицированы между двумя исходными родительскими линиями Nod+, а также между двумя наборами RIL.Три подхода NGS, включая TES, RNA-seq и GBS, применялись и сравнивались для идентификации SNP. Чтобы обобщить и сравнить различные методы анализа SNP, мы применили и сравнили два метода выравнивания (с геномом A + B или с геномом A/B) и различные конвейеры вызова и фильтрации SNP с использованием данных секвенирования. Кроме того, массив Axiom Arachis2 также использовался для генотипирования и служил платформой перекрестной проверки SNP для идентифицированных SNP.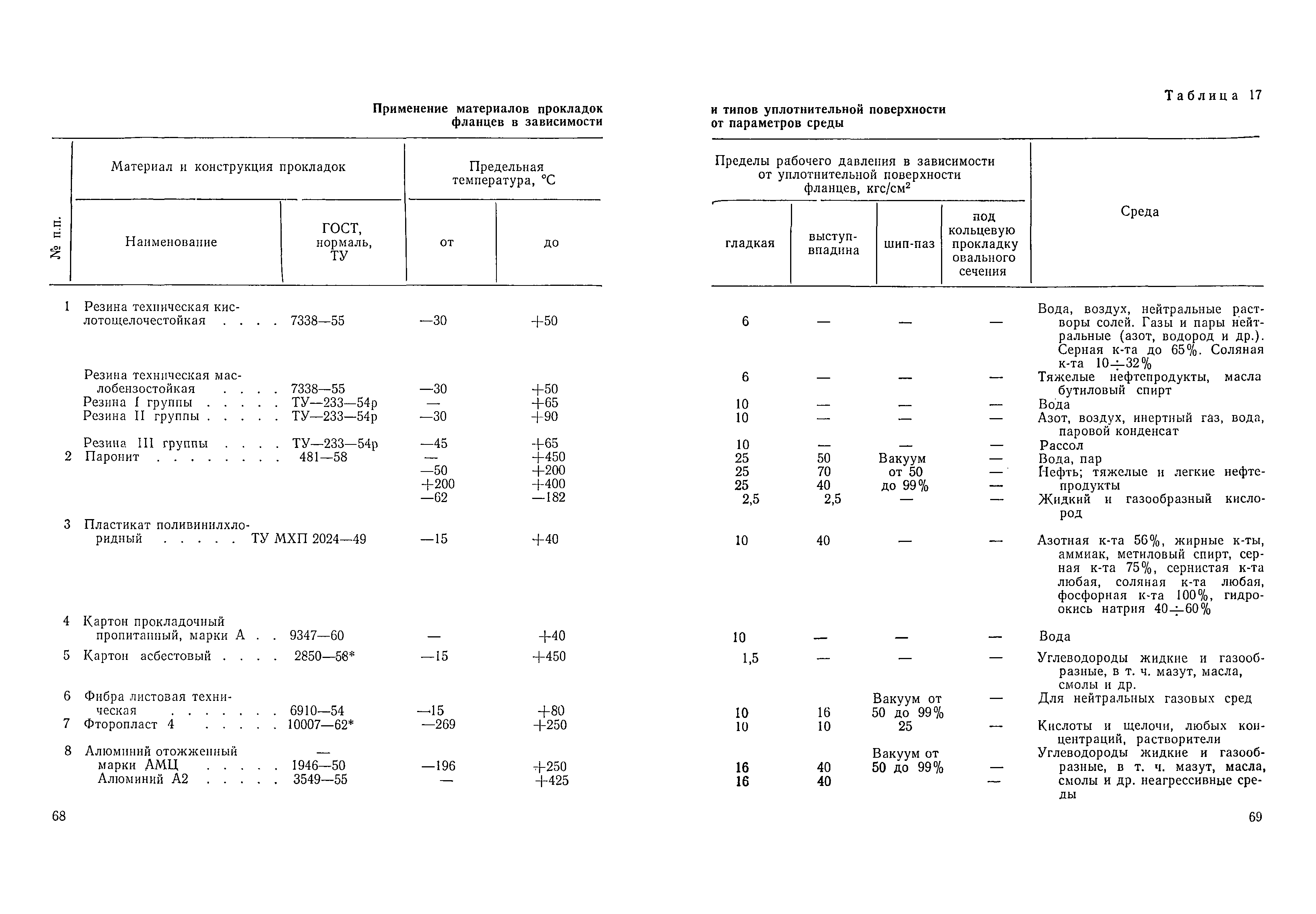 Это первое исследование, в котором сравниваются различные конвейеры вызова и фильтрации SNP для различных источников данных NGS в арахисе.Результаты и предложения этого исследования дают представление об идентификации SNP и генотипировании арахиса. Полиморфные геномные области между сестринскими RIL выявили гены-кандидаты, контролирующие клубеньки арахиса, которые будут полезны для будущих исследований генетического картирования.
Это первое исследование, в котором сравниваются различные конвейеры вызова и фильтрации SNP для различных источников данных NGS в арахисе.Результаты и предложения этого исследования дают представление об идентификации SNP и генотипировании арахиса. Полиморфные геномные области между сестринскими RIL выявили гены-кандидаты, контролирующие клубеньки арахиса, которые будут полезны для будущих исследований генетического картирования.
Материалы и методы
Растительные материалы
Два набора RIL, E4 (Nod-) и E5 (Nod+) и E6 (Nod+) и E7 (Nod-), а также их родительские линии, PI 262090 (Nod+) и UF 487A (Nod+), были включены в эта учеба.Родословная этих шести линий была представлена ранее (Peng et al., 2017a). Вкратце, два набора RIL можно проследить до двух разных линий F 6 , которые были получены в результате скрещивания между PI 262090 и UF 487A. Они также являются родительскими линиями для двух картирующих популяций F 2 (E4 × E5 и E6 × E7) для генетического картирования генов нодуляции. Морфологические и генетические характеристики RIL были описаны ранее (Peng et al., 2018). Геномную ДНК шести генотипов экстрагировали с использованием метода CTAB (Rogers and Bendich, 1994).Концентрацию и качество ДНК проверяли с помощью агарозного геля и NanoDrop.
Морфологические и генетические характеристики RIL были описаны ранее (Peng et al., 2018). Геномную ДНК шести генотипов экстрагировали с использованием метода CTAB (Rogers and Bendich, 1994).Концентрацию и качество ДНК проверяли с помощью агарозного геля и NanoDrop.
Дизайн зонда, оценка и выбор для секвенирования обогащения мишени
Для предпочтительного нацеливания на гены арахиса, потенциально связанные с образованием клубеньков, в конструкцию зонда была включена серия генов. Во-первых, были включены предполагаемые ортологичные гены, связанные с клубеньками, и гены с дифференциальной экспрессией (DEG) при заражении ризобиями из предыдущего отчета (Peng et al., 2017a) (называемые генами класса I).Для этих генов арахиса генные последовательности вместе с последовательностями 2 т.п.о. выше и 1 т.п.н. ниже были подвергнуты дизайну зонда. Для генов класса I, если в той же ортологичной группе с геном, связанным с образованием клубеньков, в модельных бобовых было более четырех генов арахиса, для последующего отбора зондов включались только четыре верхних гена (на основе оценки Blast).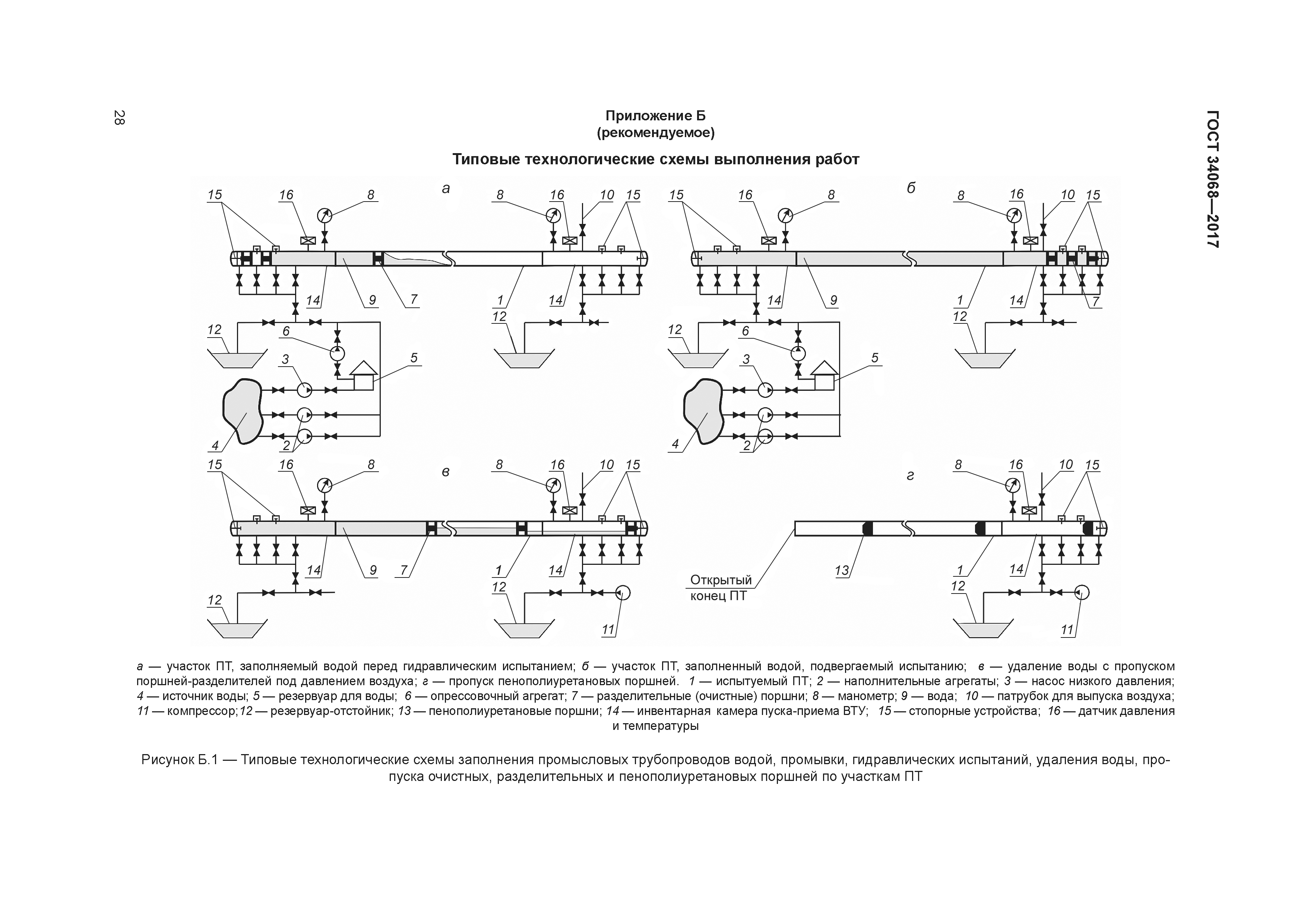 Во-вторых, для остальных генов, которые были аннотированы в геномах диплоидных предков арахиса (называемых генами класса II), для дизайна зонда использовались только последовательности, кодирующие гены.Зонды имели длину 120 п.н. и не перекрывались друг с другом. Всего в ходе предыдущего исследования транскриптома было получено 3982 гена класса I (Peng et al., 2017a). Последовательности этих генов вместе с оставшимися 74 753 моделями генов класса II в геномах диплоидных предков арахиса были представлены для разработки зондов.
Во-вторых, для остальных генов, которые были аннотированы в геномах диплоидных предков арахиса (называемых генами класса II), для дизайна зонда использовались только последовательности, кодирующие гены.Зонды имели длину 120 п.н. и не перекрывались друг с другом. Всего в ходе предыдущего исследования транскриптома было получено 3982 гена класса I (Peng et al., 2017a). Последовательности этих генов вместе с оставшимися 74 753 моделями генов класса II в геномах диплоидных предков арахиса были представлены для разработки зондов.
Зонд может захватывать или гибридизоваться с фрагментами ДНК, если они имеют сходство последовательностей друг с другом. Геномные области, имеющие сходство последовательностей с зондами, считались целевыми областями зонда.Однако эффективность захвата будет разной для целевых регионов с разным сходством. Таким образом, было исследовано количество областей-мишеней для зондов с различными порогами идентичности выравнивания, когда они были картированы в геноме. Уникальность и распространение разработанных зондов были дополнительно оценены.
Уникальность и распространение разработанных зондов были дополнительно оценены.
Чтобы оценить уникальность сконструированных зондов в геноме, последовательности зондов были сопоставлены с диплоидными геномами арахиса (A + B) с использованием Blat (Kent, 2002).Попадание определялось при отсечке: e-значение ≤ 1e-05; идентичность выравнивания = длина выравнивания × процент идентичности ≥96 (120 п.н. × 80% = 96 п.н.). Для облегчения последующего анализа данных для синтеза были выбраны в первую очередь однократные зонды. С помощью CD-HIT-EST был получен уникальный набор однократных зондов (-c 0,8 -aL 0,8 -AL 24 -aS 0,8 -AS 24 -n 5 -T 0 -r 1) (Fu et al., 2012). ). Были отобраны все однократные зонды, охватывающие гены класса I и гены устойчивости, аннотированные в геноме. Остальные однократные зонды были отобраны для обеспечения равномерного распределения по всему геному.Для этого последовательности генома разрезали на фрагменты с помощью EMBOSS (Rice et al., 2000) и из каждого фрагмента выбирали по одному зонду, исключая фрагменты, уже покрытые ранее отобранными зондами.
Синтезированные зонды были использованы для захвата фрагментов ДНК шести генотипов. Захваченные фрагменты ДНК секвенировали с использованием платформы Illumina HiSeq 3000 (считывания парных концов 100 п.н.). Дизайн зонда, синтез, подготовка библиотеки, обогащение мишени и секвенирование выполнены компанией Rapid Genomics LLC (Флорида, США).
Эффективность захвата цели и охват зондов
Для оценки областей-мишеней зонда последовательности сконструированных зондов были сопоставлены с геномами A + B с использованием Blat в соответствии с теми же критериями, что и выше. Охват чтения для целевых областей зонда был оценен. Кроме того, была исследована взаимосвязь между охватом считывания и сходством последовательностей целевых областей с зондами, что может указывать на влияние идентичности выравнивания зондов на эффективность захвата.Чтобы добиться этого, для определения попадания применялись различные пороговые значения идентичности выравнивания, в том числе 96, 90, 84, 78, 72, 66 и 60, что соответствует совпадению 80, 75, 70, 65, 60, 55 и 50%. последовательностей зондов в геном. Координаты этих попаданий в геноме были расширены на 100 п.н. в обе стороны (в файле BED), которые в дальнейшем служили целевыми областями. Bedtools v2.24.0 (intersect) использовался для оценки охвата прочтений для целевых регионов. Были использованы файлы выравнивания как для общих, так и для уникально сопоставленных ридов, созданных из BWA-mem (Li and Durbin, 2009), как описано в разделе ниже.Таким образом, всего было включено семь файлов BED целевых регионов с разными пороговыми значениями идентичности выравнивания для расчета целевой скорости и охвата прочтений.
последовательностей зондов в геном. Координаты этих попаданий в геноме были расширены на 100 п.н. в обе стороны (в файле BED), которые в дальнейшем служили целевыми областями. Bedtools v2.24.0 (intersect) использовался для оценки охвата прочтений для целевых регионов. Были использованы файлы выравнивания как для общих, так и для уникально сопоставленных ридов, созданных из BWA-mem (Li and Durbin, 2009), как описано в разделе ниже.Таким образом, всего было включено семь файлов BED целевых регионов с разными пороговыми значениями идентичности выравнивания для расчета целевой скорости и охвата прочтений.
Наборы данных RNA-seq и GBS
Данные секвенирования РНК этих шести генотипов были получены из предыдущего исследования корневого транскриптома (Peng et al., 2017a), которые были депонированы в Архиве считывания последовательностей (SRA) Национального центра биотехнологической информации (NCBI, инвентарный номер SRP093688, BioProject PRJNA354154 и BioSample SAMN06041692-SAMN06041727). Каждый генотип имел шесть библиотек кДНК, всего 36 библиотек кДНК для шести образцов. Всего для анализа было включено 403 245 464 пары прочтений (150 п.н.). Необработанные чтения были обрезаны с помощью Trimmomatic (Bolger et al., 2014).
Каждый генотип имел шесть библиотек кДНК, всего 36 библиотек кДНК для шести образцов. Всего для анализа было включено 403 245 464 пары прочтений (150 п.н.). Необработанные чтения были обрезаны с помощью Trimmomatic (Bolger et al., 2014).
Данные GBS были получены для каждого генотипа ранее, как описано Peng et al. (2017б). Рестриктазу Ape KI использовали для удаления повторяющихся участков с целью уменьшения сложности генома. Всего было получено 17 408 637 считываний с одного конца (100 п.н.) (данные депонированы в архивах считывания последовательностей в NCBI под инвентарным номером SRP154150).Необработанные чтения из данных GBS были обрезаны до 64 п.н. с использованием стеков (Catchen et al., 2013).
Чтение выравнивания, вызов SNP и фильтрация
Выравнивание проводили двумя общими методами (табл. 1). В первом методе усеченные чтения были картированы в геноме A или B (A/B) отдельно, и все сопоставленные чтения использовались для вызова SNP (M1, M4; таблица 1). В этом методе чтение, происходящее из генома B, может быть ошибочно сопоставлено с геномом A, поскольку геномы A и B очень похожи (Bertioli et al., 2016). Вызов SNP выполнялся с помощью Samtools 1.3.1 (Li et al., 2009), встроенного в конвейер SWEEP. Сгенерированные гомеологичные SNP далее использовались в качестве якоря для последующей фильтрации SNP с помощью SWEEP и инструмента машинного обучения SNP-ML (M1). Кроме того, также использовался инструмент генотипирования на основе гаплотипов HAPLOSWEEP (M4). Таким образом, M1 был определен как выравнивание с геномом A/B с использованием общего выровненного считывания и фильтрации SNP на основе SWEEP + SNP-ML и глубины. M4 был определен как выравнивание с геномом A/B с использованием общего выровненного считывания и фильтрации SNP с использованием HAPLOSWEEP (таблица 1).Во втором методе усеченные чтения были картированы на in silico конкатенированный (A + B) тетраплоидный геном (конкатенированный из диплоидных геномов), и для последующего анализа использовались только уникально картированные чтения (M2, M3, M5; таблица 1).
В этом методе чтение, происходящее из генома B, может быть ошибочно сопоставлено с геномом A, поскольку геномы A и B очень похожи (Bertioli et al., 2016). Вызов SNP выполнялся с помощью Samtools 1.3.1 (Li et al., 2009), встроенного в конвейер SWEEP. Сгенерированные гомеологичные SNP далее использовались в качестве якоря для последующей фильтрации SNP с помощью SWEEP и инструмента машинного обучения SNP-ML (M1). Кроме того, также использовался инструмент генотипирования на основе гаплотипов HAPLOSWEEP (M4). Таким образом, M1 был определен как выравнивание с геномом A/B с использованием общего выровненного считывания и фильтрации SNP на основе SWEEP + SNP-ML и глубины. M4 был определен как выравнивание с геномом A/B с использованием общего выровненного считывания и фильтрации SNP с использованием HAPLOSWEEP (таблица 1).Во втором методе усеченные чтения были картированы на in silico конкатенированный (A + B) тетраплоидный геном (конкатенированный из диплоидных геномов), и для последующего анализа использовались только уникально картированные чтения (M2, M3, M5; таблица 1). . В этом методе использовались только риды, имеющие уникальное расположение в тетраплоидном геноме (по данным выравнивателя). Вызов SNP выполняли с помощью Samtools (Li et al., 2009). Фильтрация SNP выполнялась с использованием обычной фильтрации, основанной только на глубине чтения (M2), SWEEP и SNP-ML (M3) или HAPLOSWEEP (M5).Таким образом, M2 был определен как выравнивание с геномом A + B с использованием уникально картированных прочтений и фильтрации SNP на основе глубины. M3 был определен как выравнивание с геномом A + B с использованием уникально сопоставленных прочтений и фильтрации SNP на основе SWEEP + SNP-ML и глубины. M5 был определен как выравнивание с геномом A + B с использованием уникально картированных прочтений и фильтрации SNP с использованием HAPLOSWEEP (таблица 1).
. В этом методе использовались только риды, имеющие уникальное расположение в тетраплоидном геноме (по данным выравнивателя). Вызов SNP выполняли с помощью Samtools (Li et al., 2009). Фильтрация SNP выполнялась с использованием обычной фильтрации, основанной только на глубине чтения (M2), SWEEP и SNP-ML (M3) или HAPLOSWEEP (M5).Таким образом, M2 был определен как выравнивание с геномом A + B с использованием уникально картированных прочтений и фильтрации SNP на основе глубины. M3 был определен как выравнивание с геномом A + B с использованием уникально сопоставленных прочтений и фильтрации SNP на основе SWEEP + SNP-ML и глубины. M5 был определен как выравнивание с геномом A + B с использованием уникально картированных прочтений и фильтрации SNP с использованием HAPLOSWEEP (таблица 1).
Таблица 1. Пять различных конвейеров выравнивания и фильтрации SNP.
При анализе данных TES и GBS Bowtie2/2.3.4.1 (по умолчанию –sensitive-local) использовался для выравнивания прочтений с геномами A и B отдельно (для первого метода) с последующей фильтрацией SNP, которая ранее широко применялась в арахисе (Clevenger et al. , 2017, 2018; Pandey). и др., 2017). Из-за низкой уникальной скорости отображения от Bowtie2 для выравнивания чтения (для второго метода) использовалась BWA-mem, которая применялась в нашем предыдущем отчете TES (Peng et al., 2017b). Уникально сопоставленные чтения из BWA-mem были извлечены путем фильтрации чтений с нулевым качеством отображения и тегом «XA: Z».При анализе данных секвенирования РНК как для Первого, так и для Второго методов использовался расщепленный выравниватель Tophat2.1.1 (Kim et al., 2013) с одним несоответствием в исходном файле 20 п.н. и предоставленных файлах GFF (Bertioli et al. , 2016). Уникально сопоставленные чтения были извлечены с использованием тега «NH:i:1» и качества сопоставления «50». Опция «–ultimate» использовалась в SWEEP с настройками по умолчанию для других опций. Для SNP-ML «-iM peanut_RNA» использовалось для данных TES и RNA-seq, а «-iM peanut_DNA» использовалось для данных GBS. Для HAPLOSWEEP «HAPLOSWEEP_LONGRANGE» использовался для данных TES и RNA-seq (чтения с парными концами), а «HAPLOSWEEP» использовался для данных GBS (чтения с одним концом).
, 2017, 2018; Pandey). и др., 2017). Из-за низкой уникальной скорости отображения от Bowtie2 для выравнивания чтения (для второго метода) использовалась BWA-mem, которая применялась в нашем предыдущем отчете TES (Peng et al., 2017b). Уникально сопоставленные чтения из BWA-mem были извлечены путем фильтрации чтений с нулевым качеством отображения и тегом «XA: Z».При анализе данных секвенирования РНК как для Первого, так и для Второго методов использовался расщепленный выравниватель Tophat2.1.1 (Kim et al., 2013) с одним несоответствием в исходном файле 20 п.н. и предоставленных файлах GFF (Bertioli et al. , 2016). Уникально сопоставленные чтения были извлечены с использованием тега «NH:i:1» и качества сопоставления «50». Опция «–ultimate» использовалась в SWEEP с настройками по умолчанию для других опций. Для SNP-ML «-iM peanut_RNA» использовалось для данных TES и RNA-seq, а «-iM peanut_DNA» использовалось для данных GBS. Для HAPLOSWEEP «HAPLOSWEEP_LONGRANGE» использовался для данных TES и RNA-seq (чтения с парными концами), а «HAPLOSWEEP» использовался для данных GBS (чтения с одним концом).
Наконец, SNP, вызванные из методов M1, M2 и M3, были отфильтрованы на основе глубины чтения. Гомозиготный генотип называли, если имелось не менее четырех прочтений, поддерживающих референсный или альтернативный аллель. Гетерозиготный генотип называли, если имелось не менее двух прочтений, поддерживающих эталонный и альтернативный аллели соответственно.
Генотипирование с помощью 48K Axiom
Arachis2 Массив и проверка результатов вызова SNP из конвейеров NGS Образцы ДНК шести родительских генотипов были отправлены в Affymetrix для генотипирования с использованием недавно разработанного массива 48K Axiom Arachis2 .Определение генотипа проводили, как описано ранее (Clevenger et al., 2018). Все SNP (между PI 262090 и UF 487A), идентифицированные из разных используемых конвейеров, сравнивали с результатами генотипирования из массива SNP, чтобы идентифицировать перекрывающиеся или общие SNP. Полиморфные SNP (между PI 262090 и UF 487A), идентифицированные из этих конвейеров, считались проверенными или соответствующими массиву, если они также были полиморфными в массиве и имели те же генотипы, что и генотипы, полученные из методов NGS. Впоследствии были рассчитаны показатели проверки или соответствия для пяти пайплайнов анализа SNP (M1–M5).
Впоследствии были рассчитаны показатели проверки или соответствия для пяти пайплайнов анализа SNP (M1–M5).
Результаты
Дизайн и выбор зонда для секвенирования обогащения мишени
Всего было разработано 199 673 зонда для 3 982 генов класса I и 1 678 459 зондов для 74 753 моделей генов класса II. После сопоставления последовательностей зондов с геномами (A + B) Блатом в общей сложности 230 730 зондов имели одно уникальное совпадение (идентичность выравнивания ≥96) с геномами.Чтобы избежать какой-либо избыточности из-за дублирования последовательностей генома, был применен CD-HIT-EST, и в общей сложности осталось 219 850 однократных зондов. Среди однократных зондов сначала было выбрано 20 212 зондов, соответствующих 2072 генам класса I, и 9 582 зонда, охватывающих 907 генов устойчивости (дополнительная таблица S1). Кроме того, были отобраны 824 зонда с двумя, тремя или четырьмя попаданиями в геномы, поскольку они охватывали гены, не имеющие зондов с одиночным попаданием. Это привело к тому, что в общей сложности был выбран 30 081 зонд (дополнительная таблица S1), охватывающий гены класса I и устойчивости.
Это привело к тому, что в общей сложности был выбран 30 081 зонд (дополнительная таблица S1), охватывающий гены класса I и устойчивости.
Чтобы выбрать оставшиеся зонды, охватывающие модели генов класса II, последовательности генома были нарезаны на фрагменты размером 44,3 т.п.н. с помощью EMBOSS, и всего было получено 56 296 фрагментов. Исключив 2783 фрагмента, которые уже были охвачены ранее выбранными зондами, в общей сложности 24 922 фрагмента были охвачены оставшимися зондами с одним попаданием. Таким образом, были случайным образом исключены три фрагмента и выбран один зонд из каждого из оставшихся 24 919 фрагментов, так что все отобранные зонды были практически равномерно распределены по всему геному.Наконец, для экспериментов TES было отобрано в общей сложности 55 000 зондов (дополнительная таблица S1).
Сводка статистики последовательности, обрезки и выравнивания
В среднем было 14 211 850 парных прочтений (100 п.н.) на образец, полученный из TES, 67 207 577 парных прочтений (150 п. н.) на образец из RNA-seq и 2 901 440 одноконцевых прочтений (100 п.н.) на образец из GBS (дополнительная таблица S2). После обрезки 96,89% прочтений остались для TES, 88,29% для RNA-seq и все чтения остались для GBS (прочтения урезаны до 64 п.н.).Когда обрезанные чтения были выровнены с геномом A/B (геномы A и B отдельно), в среднем общая скорость картирования составляла более 96 % с геномом A или B для TES, более 53 % для РНК-seq и более 82% для GBS. При сопоставлении с конкатенированным геномом A + B средняя скорость однозначно картированных прочтений составила 51,6% для TES, 50,26% для РНК-секвенции и 19,31% для GBS (дополнительная таблица S2). Низкая скорость уникального картирования для GBS согласовывалась с его коротким чтением (64 п.н.), используемым для выравнивания, в отличие от длины чтения 100 п.н. для TES и длины чтения 150 п.н. для RNA-seq.Определенный уровень повторяющихся последовательностей может существовать в чтениях GBS, что также может привести к низкой скорости уникального картирования.
н.) на образец из RNA-seq и 2 901 440 одноконцевых прочтений (100 п.н.) на образец из GBS (дополнительная таблица S2). После обрезки 96,89% прочтений остались для TES, 88,29% для RNA-seq и все чтения остались для GBS (прочтения урезаны до 64 п.н.).Когда обрезанные чтения были выровнены с геномом A/B (геномы A и B отдельно), в среднем общая скорость картирования составляла более 96 % с геномом A или B для TES, более 53 % для РНК-seq и более 82% для GBS. При сопоставлении с конкатенированным геномом A + B средняя скорость однозначно картированных прочтений составила 51,6% для TES, 50,26% для РНК-секвенции и 19,31% для GBS (дополнительная таблица S2). Низкая скорость уникального картирования для GBS согласовывалась с его коротким чтением (64 п.н.), используемым для выравнивания, в отличие от длины чтения 100 п.н. для TES и длины чтения 150 п.н. для RNA-seq.Определенный уровень повторяющихся последовательностей может существовать в чтениях GBS, что также может привести к низкой скорости уникального картирования.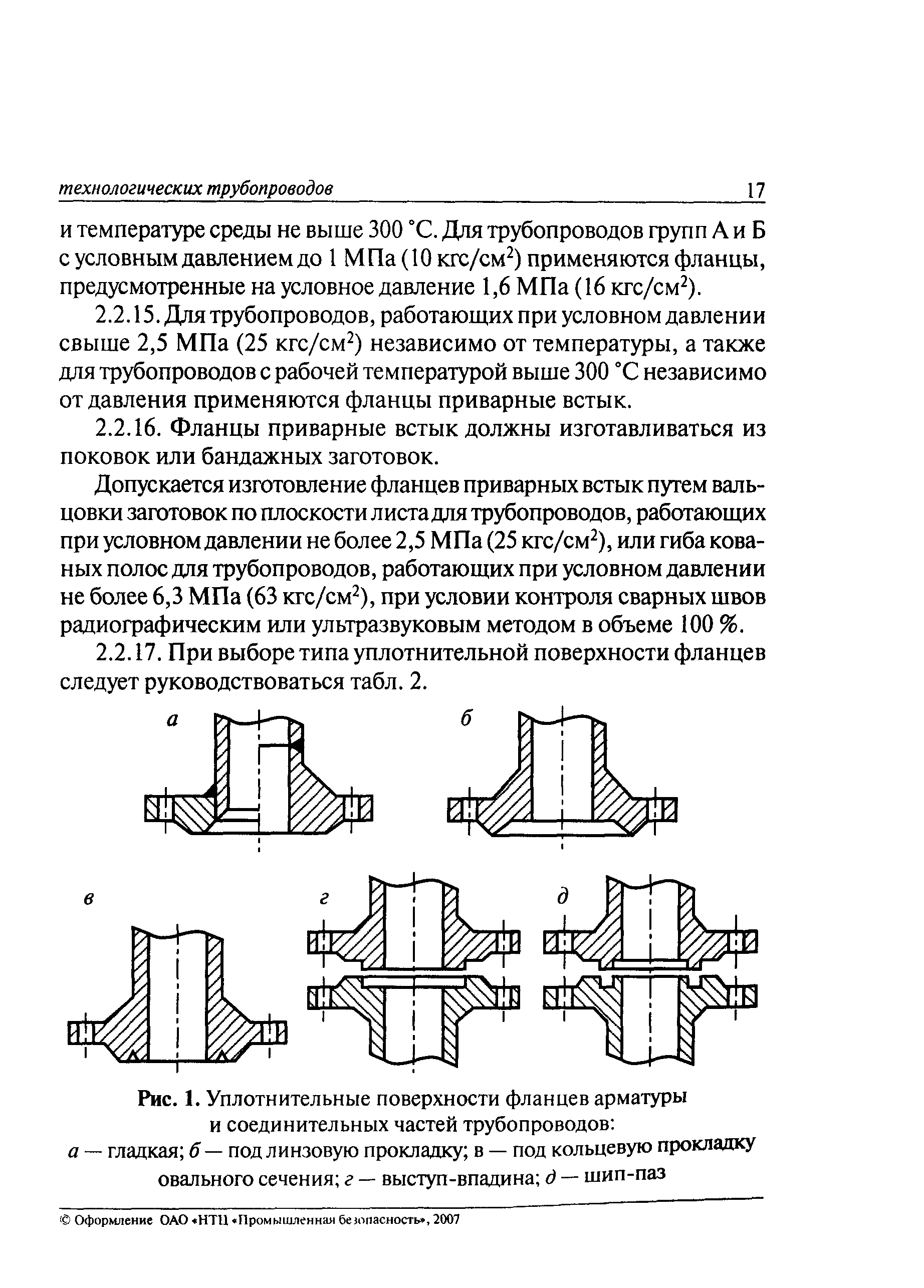 Поскольку геномы A и B были очень похожи, более короткие последовательности с меньшей вероятностью находили уникальное место при сопоставлении с геномом A + B.
Поскольку геномы A и B были очень похожи, более короткие последовательности с меньшей вероятностью находили уникальное место при сопоставлении с геномом A + B.
Оценка эффективности захвата цели и покрытия
После сопоставления последовательностей зондов с геномами при пороговом значении идентичности выравнивания ≥96 было 50 580 и 48 275 (91,96 и 87,77% из 55 002) областей-мишеней зонда, охваченных считываниями в соответствии с общими и уникально картированными считываниями соответственно (рис. 1А). .За счет уменьшения порога идентичности выравнивания стало доступно больше целевых областей, которые были охвачены считываниями. В частности, при идентичности выравнивания от 60 до ∼66 все еще оставалось 149 885 и 132 787 (79,57 и 70,49% из 188 369) целевых областей, охваченных общим выровненным прочтением и однозначно выровненным прочтением, соответственно. Средние показатели целевого сопоставления прочтений с целевыми областями с идентичностью выравнивания ≥96 составили 12,82% для всех сопоставленных прочтений и 16,28% для однозначно сопоставленных прочтений (рис. 1B).Остальные чтения были сопоставлены с целевыми областями с более низкой идентичностью выравнивания. Если рассматривать все целевые регионы с идентичностью выравнивания ≥60, средние показатели достижения цели составили 59,81 и 57,69% (рис. 1B) соответственно. Таким образом, зонды могли захватывать фрагменты ДНК даже при 50% сходстве последовательностей. Однако целевые области с более высоким сходством последовательностей с зондами имели более высокий охват чтения (рис. 1C). При пороговом значении идентичности выравнивания ≥96 целевые регионы были покрыты в среднем 29.86× и 22,05× с учетом общего и уникально сопоставленного считывания соответственно. Примечательно, что при отсечке ≥90, что соответствует сходству последовательностей ≥75%, среднее покрытие чтения составило 33,68× и 20,85× для общего и уникально картированного чтения соответственно (рис. 1C). Эффективность захвата для отсечки 90 была сравнима с эффективностью отсечки 96. Однако по мере того, как идентичность выравнивания зондов уменьшалась, средний охват прочтений, захваченных зондом, также уменьшался.
1B).Остальные чтения были сопоставлены с целевыми областями с более низкой идентичностью выравнивания. Если рассматривать все целевые регионы с идентичностью выравнивания ≥60, средние показатели достижения цели составили 59,81 и 57,69% (рис. 1B) соответственно. Таким образом, зонды могли захватывать фрагменты ДНК даже при 50% сходстве последовательностей. Однако целевые области с более высоким сходством последовательностей с зондами имели более высокий охват чтения (рис. 1C). При пороговом значении идентичности выравнивания ≥96 целевые регионы были покрыты в среднем 29.86× и 22,05× с учетом общего и уникально сопоставленного считывания соответственно. Примечательно, что при отсечке ≥90, что соответствует сходству последовательностей ≥75%, среднее покрытие чтения составило 33,68× и 20,85× для общего и уникально картированного чтения соответственно (рис. 1C). Эффективность захвата для отсечки 90 была сравнима с эффективностью отсечки 96. Однако по мере того, как идентичность выравнивания зондов уменьшалась, средний охват прочтений, захваченных зондом, также уменьшался. Таким образом, зонд может захватывать фрагменты ДНК с высокой и оптимальной эффективностью, если последовательность зонда имеет ≥75% сходства последовательностей с последовательностями фрагментов.
Таким образом, зонд может захватывать фрагменты ДНК с высокой и оптимальной эффективностью, если последовательность зонда имеет ≥75% сходства последовательностей с последовательностями фрагментов.
Рис. 1. (A) Зондирование целевых областей, (B) целевая скорость сопоставленных считываний и (C) считываний покрытия для данных секвенирования целевого обогащения.
SNP, запрашивающий данные NGS
Выравнивание, вызов SNP и фильтрация для трех различных методов NGS, данных TES, RNA-seq и GBS выполнялись с использованием пяти различных конвейеров (таблица 1). Поскольку было больше полиморфизмов между PI 262090 и UF 487A, которые были двумя исходными родительскими линиями E4, E5, E6 и E7, SNP, идентифицированные или подтвержденные между этими двумя генотипами, были суммированы и сопоставлены между пятью конвейерами для трех NGS. подходы (табл. 2).Поскольку эти шесть родительских генотипов не были включены в выборки для разработки массива Axiom Arachis2 , случайно перекрывающиеся SNP между теми, которые были идентифицированы из пяти конвейеров, и теми, которые были помещены в массив, использовались для перекрестной проверки SNP. Для данных TES наибольшее количество SNP (22 584) было от M2, за которым следуют M4 (10 157), M1 (7 540), M5 (2 694) и M3 (1 283) (таблица 2). Однако наибольшее количество гомозиготных или специфических для генома SNP было идентифицировано из M4 (10 157), что более чем в два раза больше, чем из M2 (4 438).Точно так же для данных РНК-секвенации наибольшее количество SNP было от M2 (14 684), за которым следуют M1 (1199), M4 (901), M3 (297) и M5 (288) (таблица 2). Большинство гомозиготных SNP также было идентифицировано по М4 (901), что выше, чем по М2 (787). Для данных GBS 278 SNP были идентифицированы из M4, за которыми следуют M2 (171), M1 (161), M5 (15) и M3 (9). Большинство гомозиготных SNP были вызваны из М4 (278) и М2 (37). Для всех трех источников данных M4 и M2 выявили наибольшее количество гомозиготных SNP.
Для данных TES наибольшее количество SNP (22 584) было от M2, за которым следуют M4 (10 157), M1 (7 540), M5 (2 694) и M3 (1 283) (таблица 2). Однако наибольшее количество гомозиготных или специфических для генома SNP было идентифицировано из M4 (10 157), что более чем в два раза больше, чем из M2 (4 438).Точно так же для данных РНК-секвенации наибольшее количество SNP было от M2 (14 684), за которым следуют M1 (1199), M4 (901), M3 (297) и M5 (288) (таблица 2). Большинство гомозиготных SNP также было идентифицировано по М4 (901), что выше, чем по М2 (787). Для данных GBS 278 SNP были идентифицированы из M4, за которыми следуют M2 (171), M1 (161), M5 (15) и M3 (9). Большинство гомозиготных SNP были вызваны из М4 (278) и М2 (37). Для всех трех источников данных M4 и M2 выявили наибольшее количество гомозиготных SNP.
Таблица 2. Сводка SNP между PI 262090 и UF 487A из пяти различных методов с использованием секвенирования РНК с обогащением мишени и генотипирования по данным секвенирования и степени согласованности с перекрывающимися массивами SNP.
Генотипирование с помощью Axiom
Arachis2 Массив и соответствие с методами NGS Генотипирование с использованием массива Axiom Arachis2 выявило 23 060 локусов SNP с высококачественными генотипами, требуемыми для PI 262090 и UF 487A (дополнительная таблица S3).Из 23060 локусов SNP 3531 SNP были полиморфными между PI 262090 и UF 487A, включая 2056 гомозиготных SNP и 1475 гетерозиготных SNP (дополнительная таблица S3). После сравнения SNP, идентифицированные с помощью HAPLOSWEEP, с использованием A/B или A + B в качестве эталона, всегда имели более высокую скорость проверки, чем другие методы анализа SNP, основанные на вышеупомянутых перекрывающихся SNP (81,82% для M4, 76,67% для M5). для данных ТЭС (табл. 2). Уровень проверки составил ~ 79% с учетом всех точек данных. M2 имел более низкую степень конкордантности, чем M4 и M5, но коэффициент конкордантности для гомозиготных SNP составлял 67.61%. Во всех других пайплайнах либо слишком мало SNP перекрываются с массивом, либо низкий уровень согласованности. Точно так же для данных секвенирования РНК M2, M4 и M5 выявили высокую степень согласованности с массивом SNP для гомозиготных SNP (таблица 2). Для данных GBS было слишком мало SNP из пяти пайплайнов, перекрывающихся с таковыми из массива SNP, поэтому они не были включены для сравнения.
Точно так же для данных секвенирования РНК M2, M4 и M5 выявили высокую степень согласованности с массивом SNP для гомозиготных SNP (таблица 2). Для данных GBS было слишком мало SNP из пяти пайплайнов, перекрывающихся с таковыми из массива SNP, поэтому они не были включены для сравнения.
Были специально исследованы неподтвержденные SNP среди перекрывающихся или общих локусов SNP. Для M1 большинство неподтвержденных SNP оказались полиморфными на массиве.Однако вызовы генотипа из данных последовательности не соответствовали вызовам из массива. Среди 88 перекрывающихся SNP 57 (64,77%) из них были названы гетерозиготными SNP по данным о последовательности, но гомозиготными SNP по массиву. Этот результат показал, что M1 был в состоянии идентифицировать истинные полиморфные локусы, но может не назначать правильный генотип из-за выравнивания гомеологических прочтений, в то время как специфичный для субгенома гаплотип не может быть дифференцирован. Напротив, для подходов M4 и M5, основанных на HAPLOSWEEP, большинство вызовов генотипа из данных о последовательности соответствовали таковым из массива (таблица 2). Для остальных невалидированных SNP от М4 и М5 почти все они оказались полиморфными на массиве, однако либо с PI 262090, либо с UF 487A показали гетерозиготный генотип, которые, скорее всего, были гомеологичными SNP. Эти SNP в массиве можно использовать в качестве доминирующих маркеров. Аналогично для M2, наиболее распространенный неподтвержденный тип SNP (22 из 92 перекрывающихся SNP) был классифицирован как гомозиготный SNP на основании данных о последовательности, но назван гетерозиготным SNP на основе массива.
Для остальных невалидированных SNP от М4 и М5 почти все они оказались полиморфными на массиве, однако либо с PI 262090, либо с UF 487A показали гетерозиготный генотип, которые, скорее всего, были гомеологичными SNP. Эти SNP в массиве можно использовать в качестве доминирующих маркеров. Аналогично для M2, наиболее распространенный неподтвержденный тип SNP (22 из 92 перекрывающихся SNP) был классифицирован как гомозиготный SNP на основании данных о последовательности, но назван гетерозиготным SNP на основе массива.
Сравнение различных платформ
Были дополнительно сопоставлены общие вызванные и перекрестно проверенные SNP среди пяти конвейеров от TES и RNA-seq (рис. 2).Как для данных TES, так и для данных RNA-seq небольшая доля (<50%) названных SNP была общей между M1 и M2, M2 и M4 или между M2 и M5 (рис. 2A, B). При сравнении проверенных SNP для данных TES 17 (73,91%; из 23) SNP из M5 (используя A + B в качестве эталона) уже были охвачены M4 (используя A/B в качестве эталона) (рис. 2C). ), оба из которых применяли HAPLOSWEEP. Однако лишь небольшая часть (14 из 57, 24,56%) SNP от M2 перекрывалась с M4, хотя оба показали высокую степень проверки гомозиготных SNP (рис. 2C).Это также наблюдалось для данных RNA-seq, в которых только 4 (12,12%) из 33 SNP из M2 были покрыты M4 (рис. 2D). Эти результаты показали, что M2 и M4/M5 были способны идентифицировать различные части истинных гомозиготных SNP среди существующих истинных полиморфизмов.
2C). ), оба из которых применяли HAPLOSWEEP. Однако лишь небольшая часть (14 из 57, 24,56%) SNP от M2 перекрывалась с M4, хотя оба показали высокую степень проверки гомозиготных SNP (рис. 2C).Это также наблюдалось для данных RNA-seq, в которых только 4 (12,12%) из 33 SNP из M2 были покрыты M4 (рис. 2D). Эти результаты показали, что M2 и M4/M5 были способны идентифицировать различные части истинных гомозиготных SNP среди существующих истинных полиморфизмов.
Рисунок 2. Сравнение идентифицированных и согласованных SNP среди пяти конвейеров анализа SNP для секвенирования целевого обогащения и данных секвенирования РНК. Для панелей (A,B) число снаружи показывает общее количество SNP, идентифицированных с помощью каждого метода.Для панелей (C,D) число перед «/» показывает количество проверенных SNP, число после «/» показывает количество SNP из каждого метода, которые перекрываются с массивом Axiom Arachis2 SNP.
Были сопоставлены производительность вызова SNP и характеристики трех методов NGS, а также массива Axiom Arachis2 SNP (таблица 3). TES выявил наибольшее количество гомозиготных SNP (15 947), за ними следуют массив Axiom Arachis2 (1887), RNA-seq (1633) и GBS (312) (таблица 3).Стоимость образца для TES была высокой по сравнению с другими методами, но стоимость образца для SNP была ниже, чем для RNA-seq и GBS. Однако для разработки зонда TES требовалось предварительное знание последовательностей ДНК. Самая низкая стоимость выборки на SNP была получена с массивом Axiom Arachis2 , который также требовал наименьших усилий по анализу. Все три метода NGS требовали биоинформатического анализа данных секвенирования.
TES выявил наибольшее количество гомозиготных SNP (15 947), за ними следуют массив Axiom Arachis2 (1887), RNA-seq (1633) и GBS (312) (таблица 3).Стоимость образца для TES была высокой по сравнению с другими методами, но стоимость образца для SNP была ниже, чем для RNA-seq и GBS. Однако для разработки зонда TES требовалось предварительное знание последовательностей ДНК. Самая низкая стоимость выборки на SNP была получена с массивом Axiom Arachis2 , который также требовал наименьших усилий по анализу. Все три метода NGS требовали биоинформатического анализа данных секвенирования.
Таблица 3. Сравнение секвенирования обогащения мишени, секвенирования РНК, генотипирования путем секвенирования и массива Axiom Arachis2 .
Построение графических карт, содержащих полиморфные области между E4 и E5 и E6 и E7
Среди гомозиготных SNP между PI 262090 и UF 487A из массива Axiom Arachis2 1859 (90,68%; из 2050 SNP с высококачественными генотипами) были мономорфными между E4 и E5; 1519 (74,94%; из 2027 SNP с высококачественными генотипами) были мономорфными между E6 и E7. Путем объединения отфильтрованных SNP, идентифицированных тремя методами NGS, а также SNP из массива Axiom Arachis2 , было получено в общей сложности 19 607 неперекрывающихся гомозиготных SNP между PI 262090 и UF 487A.Среди этих гомозиготных SNP в общей сложности 222 и 1200 были дополнительно получены между E4 и E5 и E6 и E7, соответственно, после фильтрации. Таким образом, они были нанесены на графические карты генотипов (рис. 3, 4). В общей сложности было получено 75 полиморфных областей генома для E4 и E5, и 512 полиморфных областей генома были получены для E6 и E7, которые в основном охватывают и уточняют те области генома, которые были выявлены с помощью маркеров SSR (Peng et al., 2018) и потенциально содержат гены. контроль клубеньков арахиса.В 75 областях-кандидатах E4 и E5 было в общей сложности 67 DEG и 26 предполагаемых ортологичных генов, связанных с клубеньками, среди которых CLE13 , ENOD16 , NFR5 и NSP2 также были DEG (дополнительная таблица).
Путем объединения отфильтрованных SNP, идентифицированных тремя методами NGS, а также SNP из массива Axiom Arachis2 , было получено в общей сложности 19 607 неперекрывающихся гомозиготных SNP между PI 262090 и UF 487A.Среди этих гомозиготных SNP в общей сложности 222 и 1200 были дополнительно получены между E4 и E5 и E6 и E7, соответственно, после фильтрации. Таким образом, они были нанесены на графические карты генотипов (рис. 3, 4). В общей сложности было получено 75 полиморфных областей генома для E4 и E5, и 512 полиморфных областей генома были получены для E6 и E7, которые в основном охватывают и уточняют те области генома, которые были выявлены с помощью маркеров SSR (Peng et al., 2018) и потенциально содержат гены. контроль клубеньков арахиса.В 75 областях-кандидатах E4 и E5 было в общей сложности 67 DEG и 26 предполагаемых ортологичных генов, связанных с клубеньками, среди которых CLE13 , ENOD16 , NFR5 и NSP2 также были DEG (дополнительная таблица).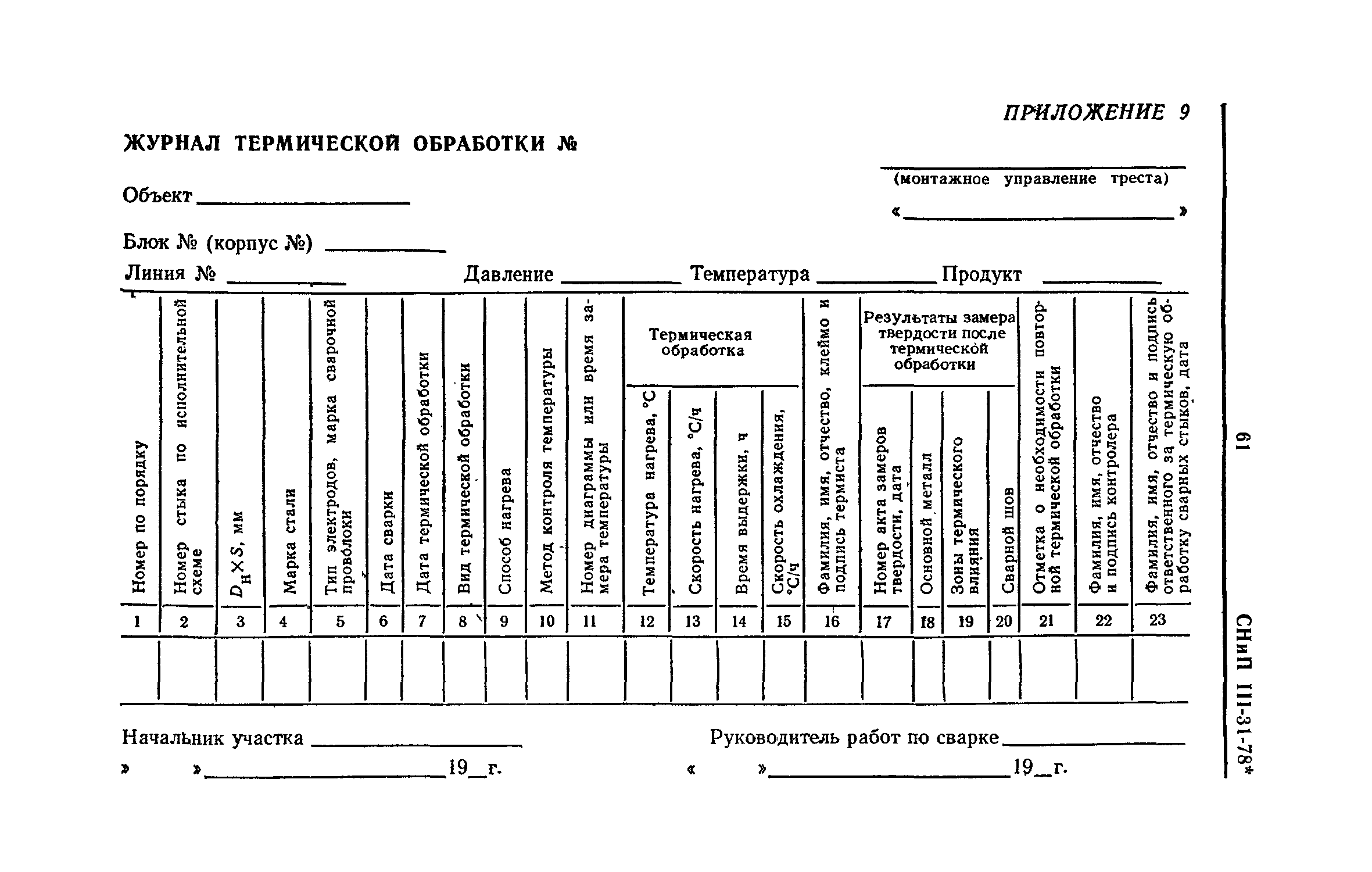 С4). В 512 областях-кандидатах E6 и E7 было в общей сложности 217 DEG и 39 предполагаемых ортологичных генов, связанных с клубеньками, среди которых CLE13 , ENOD16 и RIP1 также были DEG (дополнительная таблица S4).Эти гены могут служить генами-кандидатами, контролирующими клубеньки арахиса, для дальнейшего генетического и точного картирования.
С4). В 512 областях-кандидатах E6 и E7 было в общей сложности 217 DEG и 39 предполагаемых ортологичных генов, связанных с клубеньками, среди которых CLE13 , ENOD16 и RIP1 также были DEG (дополнительная таблица S4).Эти гены могут служить генами-кандидатами, контролирующими клубеньки арахиса, для дальнейшего генетического и точного картирования.
Рисунок 3. Графическая карта, показывающая полиморфные области генома между E4 и E5. Каждая линия представляет собой гомозиготный SNP. Каждый кружок представляет ген-кандидат.
Рисунок 4. Графическая карта, показывающая полиморфные области генома между E6 и E7. Каждая линия представляет собой гомозиготный SNP. Каждый кружок представляет ген-кандидат.
Обсуждение
В этом исследовании мы в основном сосредоточились на идентификации полиморфных областей между двумя парами сестринских RIL, E4 и E5, а также E6 и E7, которые являются почти изогенными линиями.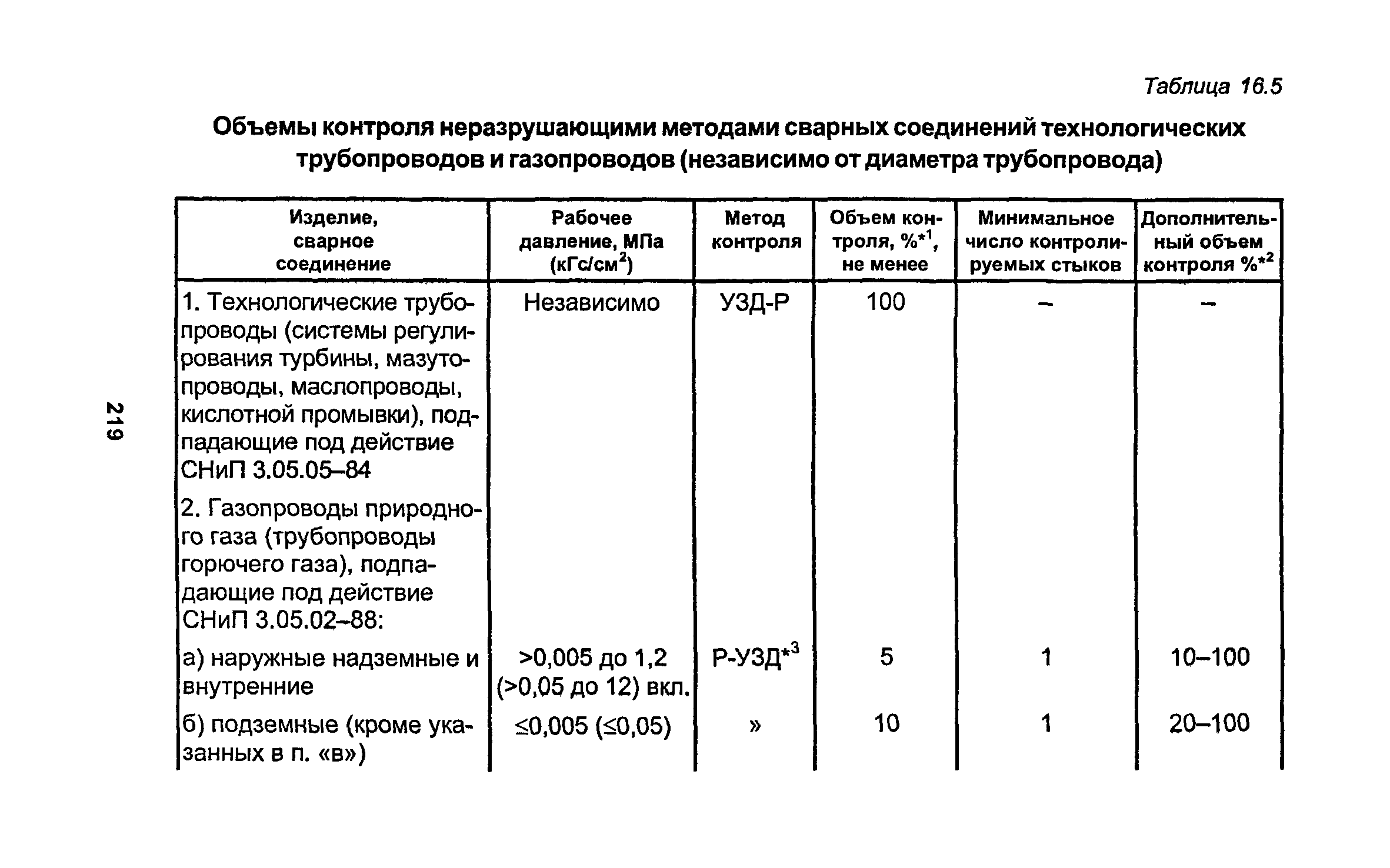 Для картирования или точного картирования генов, контролирующих образование клубеньков, полиморфные маркеры, дифференцирующие почти изогенные сестринские линии, имеют решающее значение, и их сложно разработать из-за (1) аллополиплоидной природы культивируемого арахиса и (2) почти изогенной природы двух пар. сестринских линий.Поэтому в этом исследовании мы внедрили несколько методов генотипирования SNP с поддержкой NGS и конвейеры вызова SNP для определения надежного и достаточного количества маркеров SNP.
Для картирования или точного картирования генов, контролирующих образование клубеньков, полиморфные маркеры, дифференцирующие почти изогенные сестринские линии, имеют решающее значение, и их сложно разработать из-за (1) аллополиплоидной природы культивируемого арахиса и (2) почти изогенной природы двух пар. сестринских линий.Поэтому в этом исследовании мы внедрили несколько методов генотипирования SNP с поддержкой NGS и конвейеры вызова SNP для определения надежного и достаточного количества маркеров SNP.
Одиночные нуклеотидные полиморфизмы широко используются для генотипирования благодаря нескольким благоприятным характеристикам, таким как распространенность и высокая производительность. С развитием исследований в области геномики и генетики арахиса, особенно с появлением эталонных геномов (Bertioli et al., 2016) и массивов SNP (Clevenger et al., 2017, 2018; Pandey et al., 2017), исследовательскому сообществу арахиса стало доступно больше вариантов генотипирования SNP. Для исследований генетического картирования подход WGRS теоретически может обеспечить самое высокое разрешение плотностей маркеров.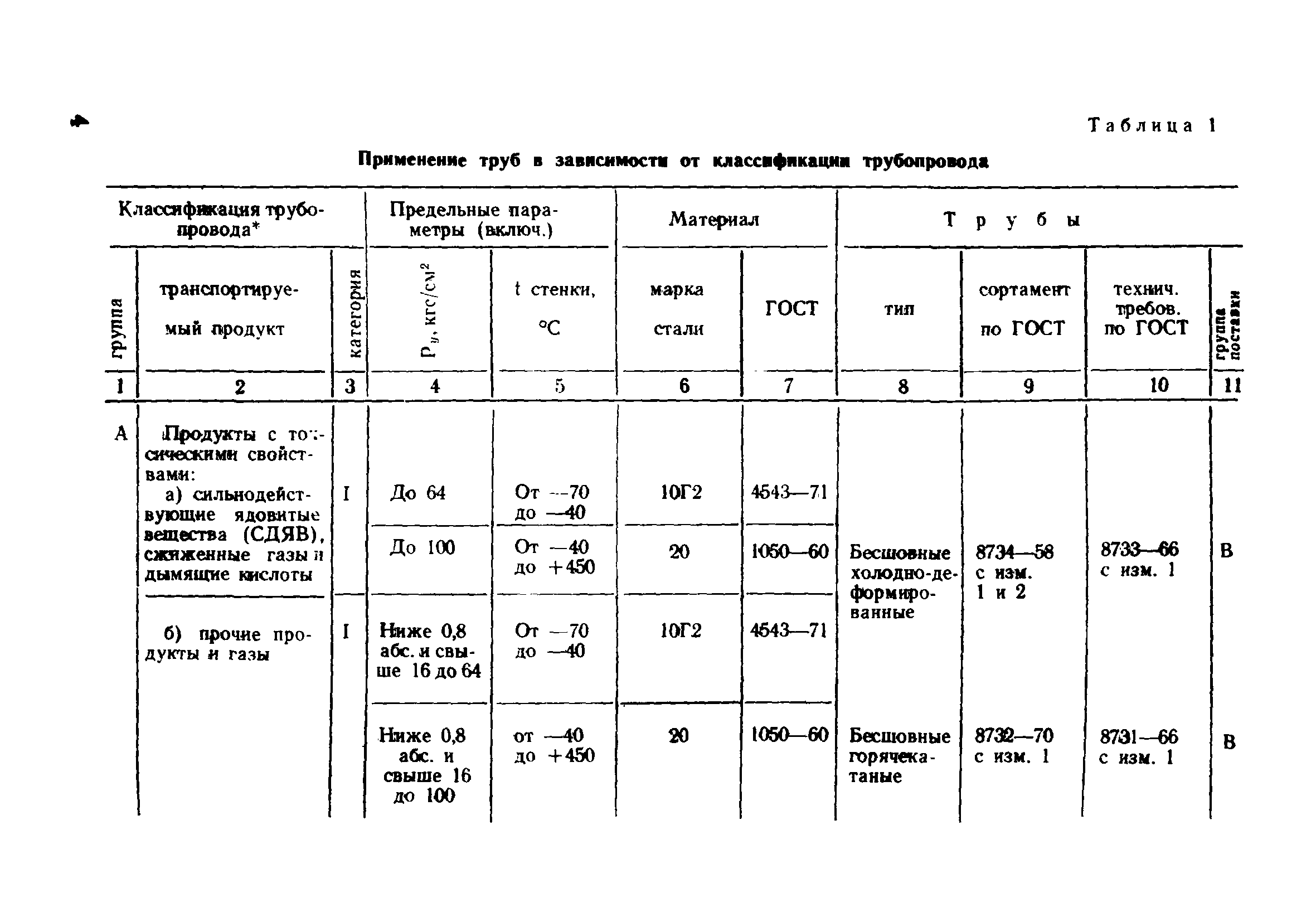 Однако для сельскохозяйственных культур, таких как арахис, с большим размером генома (∼2,7 Гб) иметь достаточно данных секвенирования, чтобы удовлетворить требования по охвату и глубине для точной идентификации SNP, все равно будет дорого. В качестве альтернативы, многочисленные подходы, такие как TES, RNA-seq и GBS, которые уменьшают сложность генома за счет секвенирования частичного генома, могут быть более экономичными, но при этом могут обеспечить приличное количество маркеров.Кроме того, массив Axiom Arachis2 (Clevenger et al., 2018) является еще одним выбором, который требует наименьших усилий по вычислительному анализу. В этом исследовании использовались шесть образцов арахиса для сравнения идентификации SNP с использованием данных секвенирования различных высокопроизводительных методов генотипирования, TES, RNA-seq, GBS, а также массива SNP. Это сравнение между различными высокопроизводительными платформами генотипирования дало представление о производительности и количестве полезных маркеров, которые могут быть созданы на каждой платформе.
Однако для сельскохозяйственных культур, таких как арахис, с большим размером генома (∼2,7 Гб) иметь достаточно данных секвенирования, чтобы удовлетворить требования по охвату и глубине для точной идентификации SNP, все равно будет дорого. В качестве альтернативы, многочисленные подходы, такие как TES, RNA-seq и GBS, которые уменьшают сложность генома за счет секвенирования частичного генома, могут быть более экономичными, но при этом могут обеспечить приличное количество маркеров.Кроме того, массив Axiom Arachis2 (Clevenger et al., 2018) является еще одним выбором, который требует наименьших усилий по вычислительному анализу. В этом исследовании использовались шесть образцов арахиса для сравнения идентификации SNP с использованием данных секвенирования различных высокопроизводительных методов генотипирования, TES, RNA-seq, GBS, а также массива SNP. Это сравнение между различными высокопроизводительными платформами генотипирования дало представление о производительности и количестве полезных маркеров, которые могут быть созданы на каждой платформе.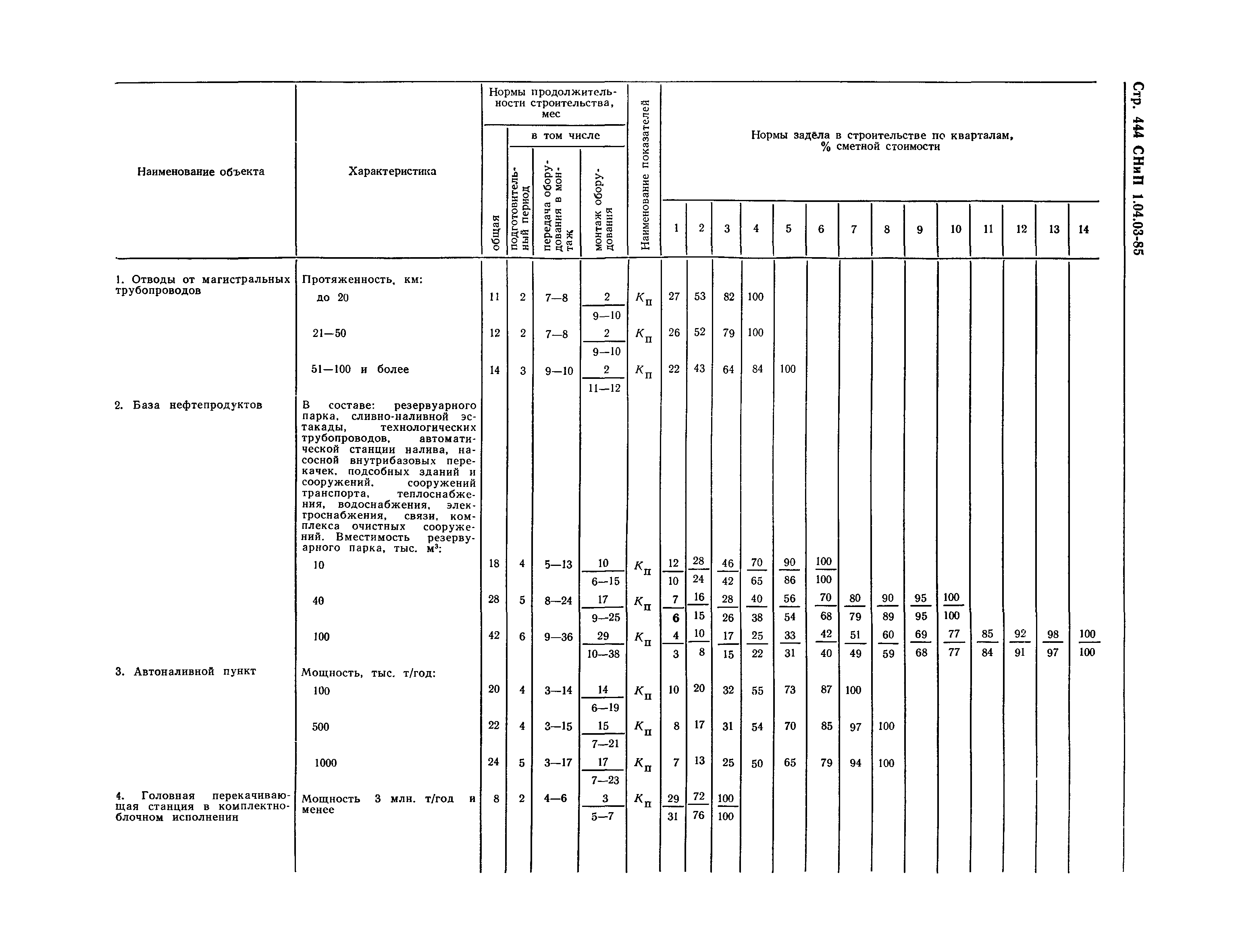 В последние несколько лет развитие маркера SNP в аллотетраплоидном арахисе с очень идентичными субгеномами было медленным из-за присутствия гомеологичных SNP (Clevenger et al., 2017). Однако с появлением таких инструментов, как SWEEP и HAPLOSWEEP, был достигнут значительный прогресс, который принесет большую пользу всему сообществу исследователей арахиса. В дополнение к этим инструментам для идентификации SNP также применялись несколько конвейеров анализа. Имея так много доступных вариантов конвейера, необходимо было сравнить их, чтобы получить лучшее представление о том, чем они отличаются друг от друга и какой из них превзошел остальные.Текущее исследование направлено на то, чтобы ответить на эти вопросы, применяя различные методы выравнивания, вызова SNP и фильтрации с различными подходами к секвенированию для идентификации SNP. Кроме того, полученные SNP выявили полиморфные геномные области между сестринскими RIL, которые могут сузить области-кандидаты, несущие гены, контролирующие образование клубеньков в арахисе, и, вероятно, облегчить будущее генетическое картирование и точное картирование генов клубеньков в арахисе.
В последние несколько лет развитие маркера SNP в аллотетраплоидном арахисе с очень идентичными субгеномами было медленным из-за присутствия гомеологичных SNP (Clevenger et al., 2017). Однако с появлением таких инструментов, как SWEEP и HAPLOSWEEP, был достигнут значительный прогресс, который принесет большую пользу всему сообществу исследователей арахиса. В дополнение к этим инструментам для идентификации SNP также применялись несколько конвейеров анализа. Имея так много доступных вариантов конвейера, необходимо было сравнить их, чтобы получить лучшее представление о том, чем они отличаются друг от друга и какой из них превзошел остальные.Текущее исследование направлено на то, чтобы ответить на эти вопросы, применяя различные методы выравнивания, вызова SNP и фильтрации с различными подходами к секвенированию для идентификации SNP. Кроме того, полученные SNP выявили полиморфные геномные области между сестринскими RIL, которые могут сузить области-кандидаты, несущие гены, контролирующие образование клубеньков в арахисе, и, вероятно, облегчить будущее генетическое картирование и точное картирование генов клубеньков в арахисе.
Секвенирование целевого обогащения
В отличие от RNA-seq и GBS, которые сосредотачиваются на областях гена или областях, окружающих сайт рестрикции, TES смог сосредоточиться на интересующих генах или геномных областях.В этом подходе фрагменты ДНК, захваченные специально разработанными зондами на основе гомологии последовательностей, были секвенированы. Исследователи могут предпочтительно разрабатывать зонды, охватывающие интересующие гены. TES впервые был применен к арахису с использованием зондов, сконструированных из экспрессированных тегов последовательности, в качестве источника последовательности для дизайна зонда (Peng et al., 2017b). В текущем исследовании для разработки зонда использовались эталонные геномы двух диплоидных предков культивируемого арахиса. Чтобы нацелиться на гены, связанные с симбиозом и устойчивостью к болезням в арахисе, было разработано в общей сложности 20 212 зондов, чтобы охватить все предполагаемые гены, связанные с клубеньками, и 9 582 зонда, чтобы охватить гены устойчивости. Остальные ∼24 тыс. зондов были отобраны для равномерного распределения по всему геному. Таким образом, общая плотность зондов составляла ~49 Кб/зонд, учитывая размер генома арахиса 2,7 Гб. Из 78 574 моделей генов арахиса 26 653 (33,9%) были помечены этим набором зондов. Этот набор зондов TES был бы полезен не только для картирования генов, связанных с образованием клубеньков или устойчивостью к болезням, но также для анализа геномных ассоциаций любых признаков с учетом плотности зондов и охвата.
Остальные ∼24 тыс. зондов были отобраны для равномерного распределения по всему геному. Таким образом, общая плотность зондов составляла ~49 Кб/зонд, учитывая размер генома арахиса 2,7 Гб. Из 78 574 моделей генов арахиса 26 653 (33,9%) были помечены этим набором зондов. Этот набор зондов TES был бы полезен не только для картирования генов, связанных с образованием клубеньков или устойчивостью к болезням, но также для анализа геномных ассоциаций любых признаков с учетом плотности зондов и охвата.
В процессе выбора зонда предпочтительно были выбраны зонды с одним попаданием, что привело к тому, что средний коэффициент уникального сопоставления пяти образцов составил 51.60%, что намного выше, чем в нашем предыдущем отчете (22,55%; Peng et al., 2017b). Кроме того, 91,96% целевых областей текущего набора зондов были покрыты считываниями со средней глубиной 29,86×, что также было намного выше, чем в нашем предыдущем отчете (средняя глубина <20× с учетом 90% целевых областей; Peng et al. ., 2017б). Таким образом, использование уникальных попаданий зондов в геном имеет решающее значение для повышения скорости однозначно картированных прочтений и глубины последовательностей, захваченных набором зондов. Основываясь на наших данных, зонды могут быть очень эффективными при захвате фрагментов ДНК, когда они имеют не менее 75% сходства последовательности с целевыми фрагментами (рис. 1C).Поэтому при применении TES следует учитывать, что нецелевой захват будет характерен именно для видов с близкородственными геномами или дублированными участками генома.
., 2017б). Таким образом, использование уникальных попаданий зондов в геном имеет решающее значение для повышения скорости однозначно картированных прочтений и глубины последовательностей, захваченных набором зондов. Основываясь на наших данных, зонды могут быть очень эффективными при захвате фрагментов ДНК, когда они имеют не менее 75% сходства последовательности с целевыми фрагментами (рис. 1C).Поэтому при применении TES следует учитывать, что нецелевой захват будет характерен именно для видов с близкородственными геномами или дублированными участками генома.
Сравнение различных подходов NGS и аксиомы
Arachis2 Массив Три источника данных NGS и массив Axiom Arachis2 определили различное количество SNP между PI 262090 и UF 487A. Принимая во внимание только гомозиготные SNP, TES выявил наибольшее количество SNP, за которым следуют массив SNP, RNA-seq и GBS (таблица 3).Это можно объяснить с нескольких точек зрения. Во-первых, поскольку TES фокусируется на геномных последовательностях, ожидается больше полиморфизмов, чем у RNA-seq, представляющих консервативные области транскрибируемых генов.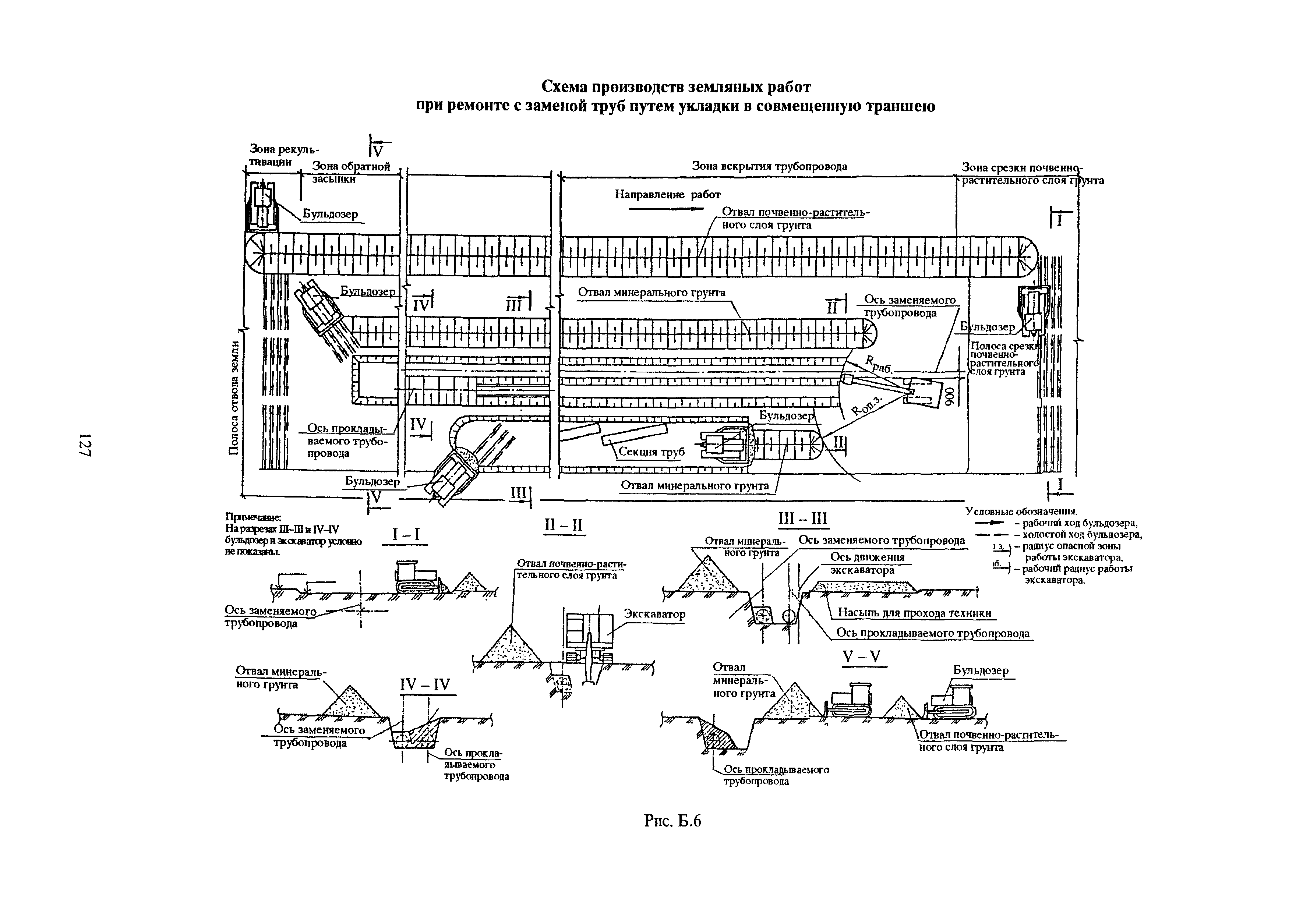 Небольшое количество SNP от GBS можно объяснить низким охватом полученных данных секвенирования. Поскольку между PI 262090 и UF 487A было получено только 2056 гомозиготных SNP, полученных из массива SNP, и еще меньше SNP для E4 и E5 и E6 и E7, массив Axiom Arachis2 SNP может не подходить для будущего генотипирования картирующих популяций. с E4 и E5 и E6 и E7 в качестве родительских линий.TES можно рассматривать как выбор из-за большого количества обнаруженных полиморфизмов. Кроме того, стоимость выборки в расчете на SNP для TES все еще низка по сравнению с другими методами NGS и сравнима с выборкой для массива Axiom Arachis2 SNP.
Небольшое количество SNP от GBS можно объяснить низким охватом полученных данных секвенирования. Поскольку между PI 262090 и UF 487A было получено только 2056 гомозиготных SNP, полученных из массива SNP, и еще меньше SNP для E4 и E5 и E6 и E7, массив Axiom Arachis2 SNP может не подходить для будущего генотипирования картирующих популяций. с E4 и E5 и E6 и E7 в качестве родительских линий.TES можно рассматривать как выбор из-за большого количества обнаруженных полиморфизмов. Кроме того, стоимость выборки в расчете на SNP для TES все еще низка по сравнению с другими методами NGS и сравнима с выборкой для массива Axiom Arachis2 SNP.
Сравнение различных конвейеров анализа SNP
По результатам сравнения пяти различных способов вызова арахисового SNP можно сделать несколько выводов. (1) Коэффициент соответствия гетерозиготных SNP всегда был низким между TES и RNA-seq.Это может быть вызвано ложноположительными SNP, полученными из-за смещения прочтений из гомеологических областей генома. (2) выравнивание с геномом A/B с последующей фильтрацией SWEEP и SNP-ML (M1) выявило значительно меньшую долю гомозиготных SNP, чем выравнивание с геномом A + B с последующей традиционной фильтрацией (M2), а HAPLOSWEEP приближается к M4, и М5. Поскольку SWEEP не смог дифференцировать гаплотипы, используя геном A/B в качестве эталона, многие истинные гомозиготные SNP можно было назвать гетерозиготными SNP из-за несовпадения.(3) M2 выявил достойную степень согласованности (67,61%) гомозиготных SNP и смог идентифицировать новые и истинные полиморфизмы, которые не были обнаружены с помощью подхода HAPLOSWEEP. (4) При использовании HAPLOSWEEP выравнивание с геномом A/B (M4) выявило больше гомозиготных SNP, чем выравнивание с геномом A + B (M5), однако M5 также могло идентифицировать новые и истинные полиморфизмы, которые не были охвачены M4. Таким образом, ни один из описанных выше конвейеров не может охватить все возможные полиморфизмы между генотипами. Однако лучшим вариантом среди пяти конвейеров анализа было выравнивание считываний с геномом A/B с последующим HAPLOSWEEP, который может дать наибольшее количество гомозиготных SNP с высокой степенью согласованности с массивом SNP, аналогично скорости, указанной в исследовании.
(2) выравнивание с геномом A/B с последующей фильтрацией SWEEP и SNP-ML (M1) выявило значительно меньшую долю гомозиготных SNP, чем выравнивание с геномом A + B с последующей традиционной фильтрацией (M2), а HAPLOSWEEP приближается к M4, и М5. Поскольку SWEEP не смог дифференцировать гаплотипы, используя геном A/B в качестве эталона, многие истинные гомозиготные SNP можно было назвать гетерозиготными SNP из-за несовпадения.(3) M2 выявил достойную степень согласованности (67,61%) гомозиготных SNP и смог идентифицировать новые и истинные полиморфизмы, которые не были обнаружены с помощью подхода HAPLOSWEEP. (4) При использовании HAPLOSWEEP выравнивание с геномом A/B (M4) выявило больше гомозиготных SNP, чем выравнивание с геномом A + B (M5), однако M5 также могло идентифицировать новые и истинные полиморфизмы, которые не были охвачены M4. Таким образом, ни один из описанных выше конвейеров не может охватить все возможные полиморфизмы между генотипами. Однако лучшим вариантом среди пяти конвейеров анализа было выравнивание считываний с геномом A/B с последующим HAPLOSWEEP, который может дать наибольшее количество гомозиготных SNP с высокой степенью согласованности с массивом SNP, аналогично скорости, указанной в исследовании. недавнее исследование (74%) (Clevenger et al., 2018). В качестве альтернативы, лучшим выбором было бы применение нескольких конвейеров для получения неизбыточных SNP. Например, методы M2 и M4 могут дополнять друг друга и давать большее количество гомозиготных SNP, если оба метода применяются для анализа.
недавнее исследование (74%) (Clevenger et al., 2018). В качестве альтернативы, лучшим выбором было бы применение нескольких конвейеров для получения неизбыточных SNP. Например, методы M2 и M4 могут дополнять друг друга и давать большее количество гомозиготных SNP, если оба метода применяются для анализа.
В этом исследовании мы использовали конкатенированные геномы A + B из диплоидных видов дикого арахиса (Bertioli et al., 2016) в качестве эталона для определения SNP вместо использования недавно опубликованных тетраплоидных геномов (Bertioli et al., 2019; Zhuang и др., 2019).Одной из наших основных целей в этом исследовании было сравнить возможности вызова SNP с использованием различных конвейеров и платформ NGS, чтобы обнаружить максимальное количество SNP в культивируемом арахисе. Это сравнение было бы надежным, если бы для сравнения различных платформ или конвейеров использовался один и тот же эталон, а SNP между эталоном и всеми чтениями были отфильтрованы. Диплоидный и тетраплоидный геномы были очень похожи (Bertioli et al. , 2019; Zhuang et al., 2019), поэтому использование любого генома в качестве эталона не изменит основных результатов этого исследования.В частности, массив Arachis2 SNP, инструмент, используемый для перекрестной проверки вызовов SNP, был разработан на основе диплоидного генома, и зонды, разработанные для TES, также относились к диплоидным геномам. Поэтому в этом исследовании для выравнивания использовался конкатенированный геном A + B из диплоидного диплоидного арахиса, чтобы добиться хорошей согласованности в сравнении.
, 2019; Zhuang et al., 2019), поэтому использование любого генома в качестве эталона не изменит основных результатов этого исследования.В частности, массив Arachis2 SNP, инструмент, используемый для перекрестной проверки вызовов SNP, был разработан на основе диплоидного генома, и зонды, разработанные для TES, также относились к диплоидным геномам. Поэтому в этом исследовании для выравнивания использовался конкатенированный геном A + B из диплоидного диплоидного арахиса, чтобы добиться хорошей согласованности в сравнении.
Геномные области-кандидаты, контролирующие клубеньки арахиса
Два набора сестринских RIL, использованных в этом исследовании, были отобраны в поколении F 6 , полученном в результате скрещивания PI 262090 и UF 487A (Peng et al., 2017а). RIL Nod+ и Nod-, особенно для E4 и E5, были очень идентичными. Воспользовавшись почти изогенной природой между двумя парами сестринских RIL, одна из которых образует узелки, а другая не образует узелков, мы предположили, что полиморфные области между сестринскими RIL должны содержать любые потенциальные гены-кандидаты, контролирующие нодуляцию.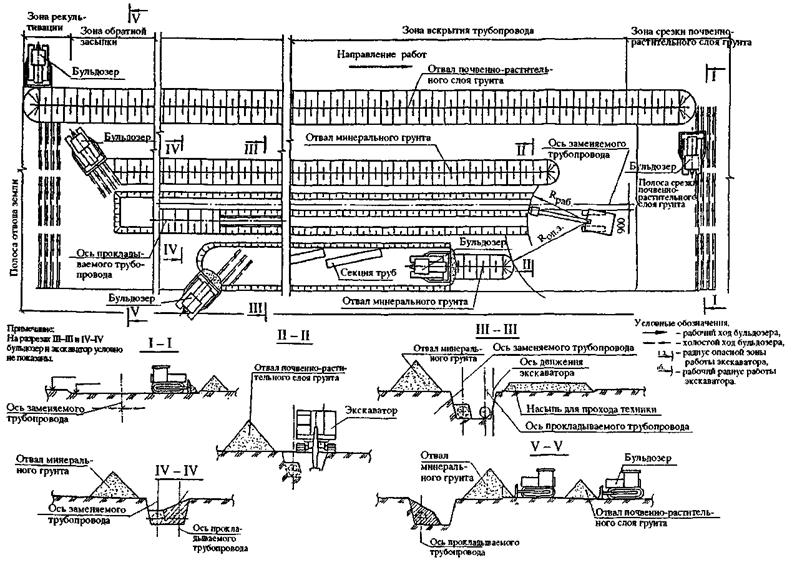 В этом исследовании, чтобы идентифицировать высоконадежные гомозиготные SNP между RIL, только гомозиготные SNP, полиморфные между PI 262090 и UF 487A, а также между RIL, были включены как высоконадежные SNP и были помещены на графические карты.Графический генотип этих двух пар RIL позволил нам визуализировать полиморфные области генома, содержащие гены-кандидаты. Полиморфные области на графических картах генотипов могут служить руководством для будущего генетического картирования генов клубеньков у арахиса, хотя эти области были довольно большими и содержали большое количество генов, поскольку в текущем исследовании еще не применялись стратегии картирования и точного картирования. Мы специально перечислили DEG, участвующие в нодуляции, и любые ортологи генов, связанных с клубеньками, в качестве кандидатов, впоследствии получив относительно большое количество кандидатов в геноме.Это большое количество генов-кандидатов было получено в результате предварительных сравнений между двумя парами почти изогенных RIL.
В этом исследовании, чтобы идентифицировать высоконадежные гомозиготные SNP между RIL, только гомозиготные SNP, полиморфные между PI 262090 и UF 487A, а также между RIL, были включены как высоконадежные SNP и были помещены на графические карты.Графический генотип этих двух пар RIL позволил нам визуализировать полиморфные области генома, содержащие гены-кандидаты. Полиморфные области на графических картах генотипов могут служить руководством для будущего генетического картирования генов клубеньков у арахиса, хотя эти области были довольно большими и содержали большое количество генов, поскольку в текущем исследовании еще не применялись стратегии картирования и точного картирования. Мы специально перечислили DEG, участвующие в нодуляции, и любые ортологи генов, связанных с клубеньками, в качестве кандидатов, впоследствии получив относительно большое количество кандидатов в геноме.Это большое количество генов-кандидатов было получено в результате предварительных сравнений между двумя парами почти изогенных RIL.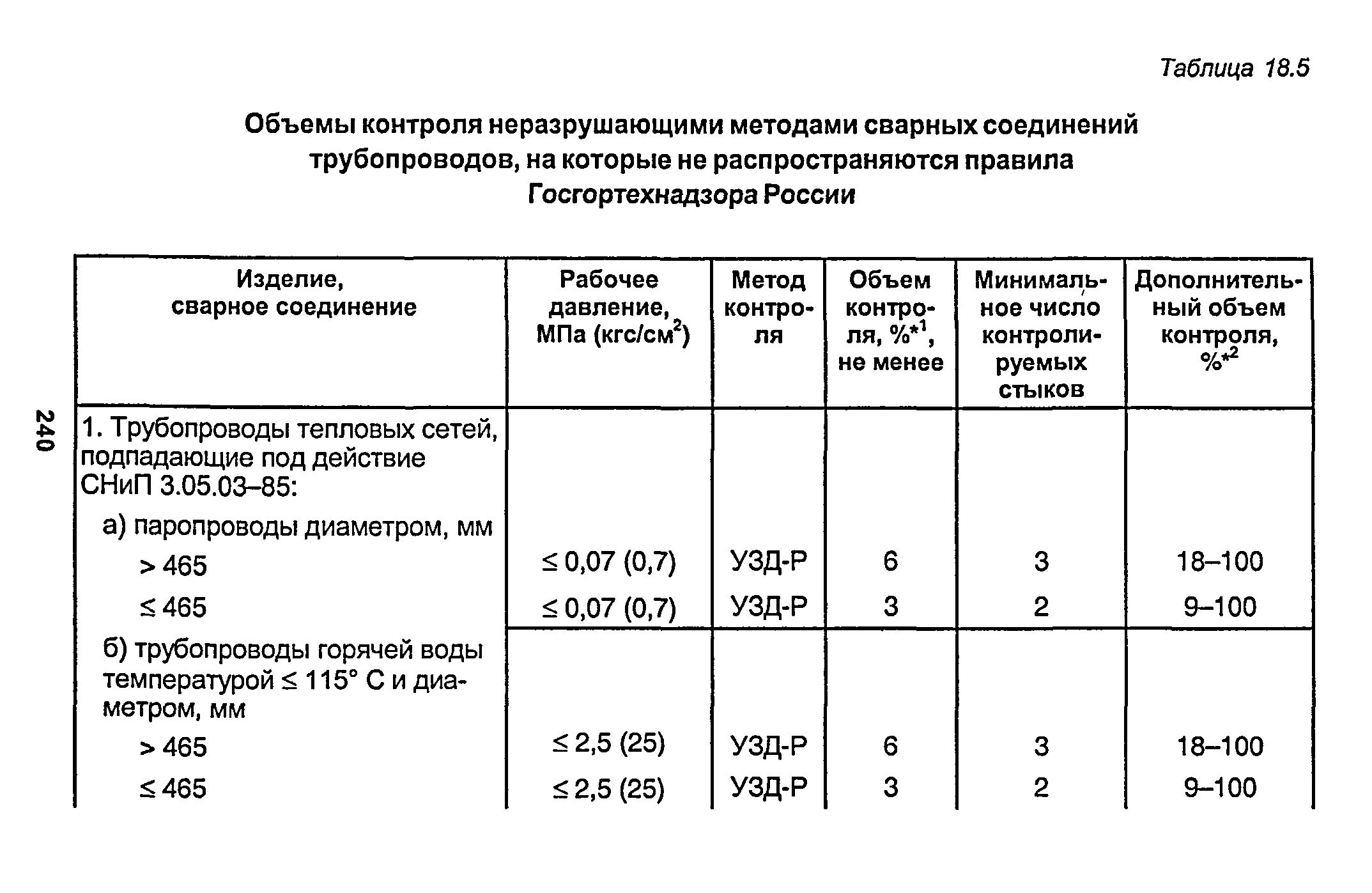 Дальнейшее картирование и стратегии точного картирования должны быть применены, чтобы сузить и точно определить гены, вызывающие отсутствие клубеньков в наших линиях без клубеньков, что будет проведено в другом исследовании.
Дальнейшее картирование и стратегии точного картирования должны быть применены, чтобы сузить и точно определить гены, вызывающие отсутствие клубеньков в наших линиях без клубеньков, что будет проведено в другом исследовании.
Заключение
На основании результатов этого исследования было сделано несколько предложений для будущих исследований по идентификации SNP в арахисе. SNP, включенные в массив Axiom Arachis2 , были в основном обнаружены в 21 генотипе арахиса, что может быть недостаточно репрезентативным, чтобы охватить все полиморфизмы генома.Массив Axiom Arachis2 был бы хорошим выбором для генотипирования популяций, созданных на основе генотипов, использованных для первоначального обнаружения SNP, или связанных с ними. Однако, если подлежащие генотипированию популяции не связаны с исходными генотипами для разработки массива Axiom Arachis2 , следует рассмотреть другие подходы NGS. Если необходимо сосредоточить внимание на интересующих генах или геномных областях, предпочтительно следует рассмотреть TES, поскольку потенциальные области-кандидаты могут быть специально включены для идентификации SNP.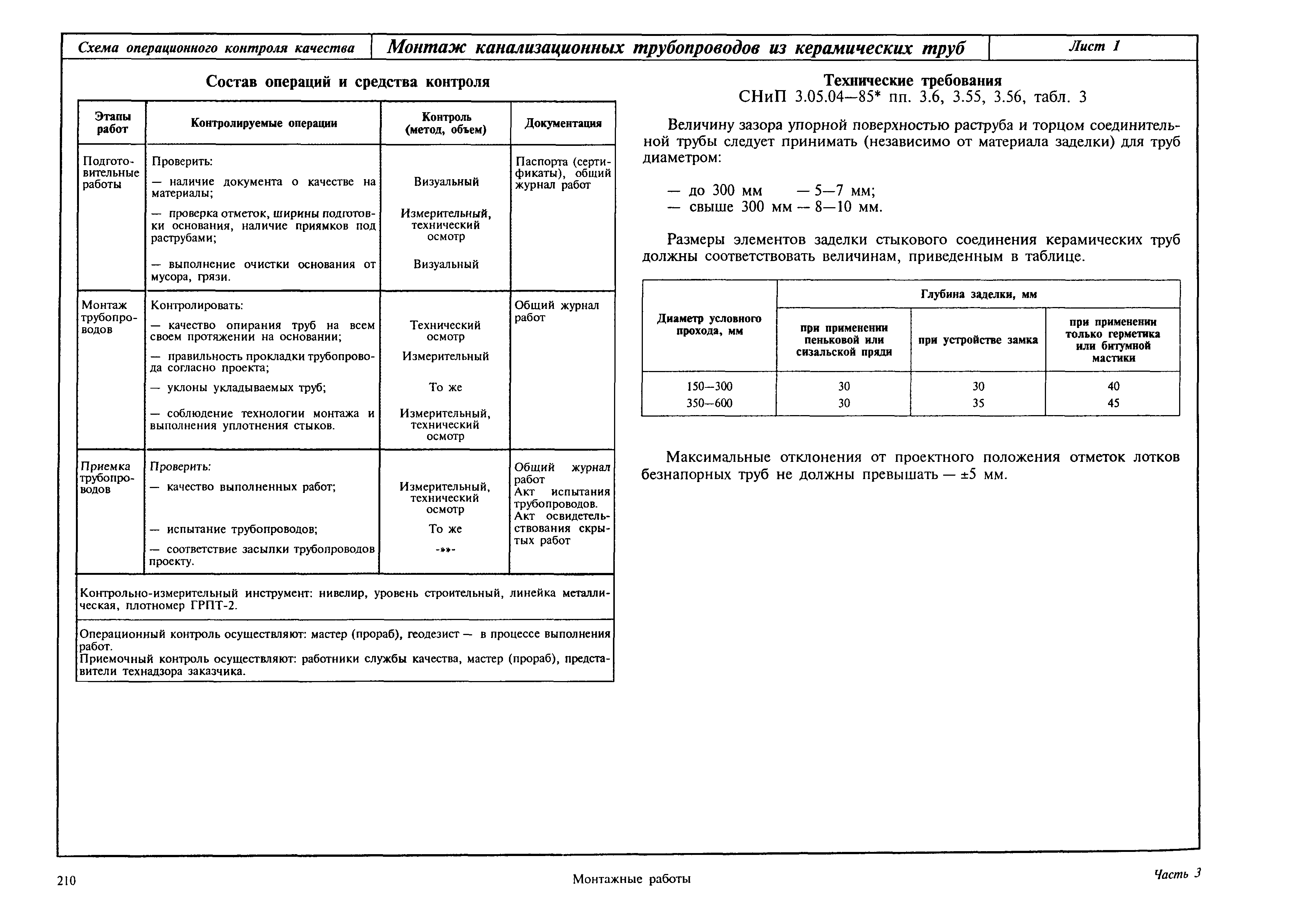 Среди конвейеров вызова SNP, которые будут использоваться для анализа данных NGS, наиболее эффективным конвейером является выравнивание считываний с геномом A/B с последующей фильтрацией SNP с использованием HAPLOSWEEP. Чтобы идентифицировать большее количество истинных гомозиготных SNP, другие конвейеры, такие как выравнивание генома A + B с традиционной фильтрацией SNP, могут быть объединены с HAPLOSWEEP.
Среди конвейеров вызова SNP, которые будут использоваться для анализа данных NGS, наиболее эффективным конвейером является выравнивание считываний с геномом A/B с последующей фильтрацией SNP с использованием HAPLOSWEEP. Чтобы идентифицировать большее количество истинных гомозиготных SNP, другие конвейеры, такие как выравнивание генома A + B с традиционной фильтрацией SNP, могут быть объединены с HAPLOSWEEP.
Заявление о доступности данных
Наборы данных, созданные для этого исследования, можно найти в NCBI SRP093688, BioProject PRJNA354154, BioSample SAMN06041692–SAMN06041727 и NCBI SRP154150.
Вклад авторов
JW задумал эксперименты и обеспечил финансирование. З.П. проводил эксперименты. ZP и ZZ проанализировали данные и составили рукопись. JC и DP помогли с анализом данных. YC и PO-A предоставили данные массива SNP. Все авторы прочитали и одобрили проект.
Финансирование
Эта работа финансировалась Фондом ранней карьеры, предоставленным Институтом пищевых и сельскохозяйственных наук (IFAS), Университетом Флориды, Национальным советом по арахису и Ассоциацией производителей арахиса Флориды. Публикация этой статьи частично финансировалась Издательским фондом открытого доступа Университета Флориды.
Публикация этой статьи частично финансировалась Издательским фондом открытого доступа Университета Флориды.
Конфликт интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могли бы быть истолкованы как потенциальный конфликт интересов.
Дополнительный материал
Дополнительный материал к этой статье можно найти в Интернете по адресу: https://www.frontiersin.org/articles/10.3389/fgene.2020.00222/полный#дополнительный материал
Ссылки
Агарвал Г., Клевенджер Дж., Панди М.К., Ван Х., Шасидхар Ю., Чу Ю. и др. (2018). Генетическая карта высокой плотности с использованием ресеквенирования всего генома для точного картирования и открытия генов-кандидатов для устойчивости к болезням у арахиса. Завод Биотехнолог. J. 16, 1954–1967. doi: 10.1111/pbi.12930
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Бертиоли, Д. Дж., Кэннон, С. Б., Френике, Л., Huang, G., Farmer, A.D., Cannon, E.K., et al. (2016). Последовательности генома Arachis duranensis и Arachis ipaensis , диплоидных предков культивируемого арахиса. Нац. Жене. 48, 438–446. doi: 10.1038/ng.3517
Дж., Кэннон, С. Б., Френике, Л., Huang, G., Farmer, A.D., Cannon, E.K., et al. (2016). Последовательности генома Arachis duranensis и Arachis ipaensis , диплоидных предков культивируемого арахиса. Нац. Жене. 48, 438–446. doi: 10.1038/ng.3517
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Бертиоли Д. Дж., Дженкинс Дж., Клевенджер Дж., Дудченко О., Гао Д., Сейджо Г. и др. (2019). Последовательность генома сегментарного аллотетраплоидного арахиса Arachis hypogaea . Нац. Жене. 51, 877–884. doi: 10.1038/s41588-019-0405-z
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Catchen, J., Hohenlohe, P.A., Bassham, S., Amores, A., and Cresko, W.A. (2013). Стеки: набор инструментов для анализа популяционной геномики. Мол. Экол. 22, 3124–3140. doi: 10.1111/mec.12354
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Чопра Р., Буроу Г.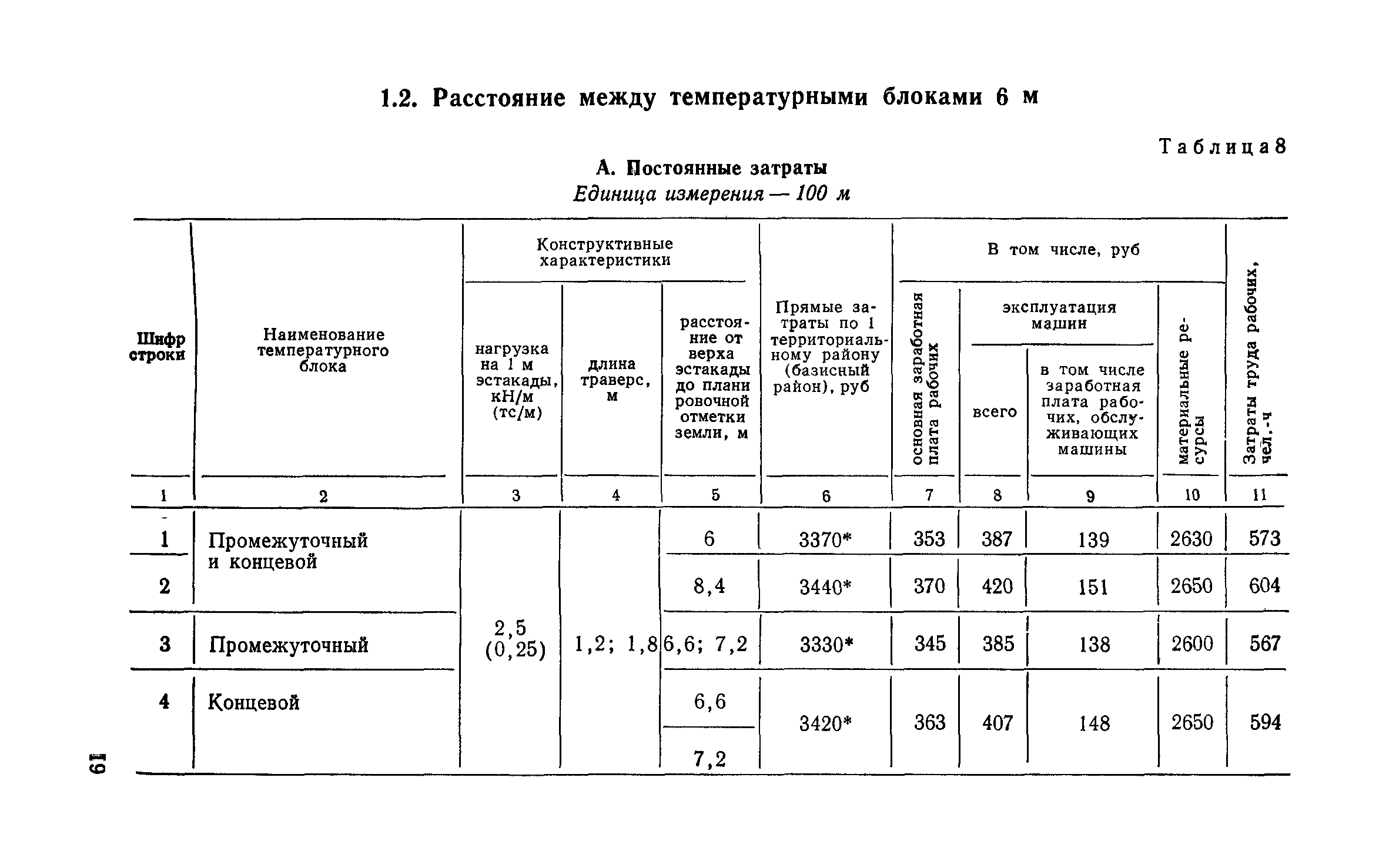 , Симпсон С. Э., Чагоя Дж., Мадж Дж. и Буроу М.Д. (2016). Секвенирование транскриптомов различных диких и культивируемых видов арахиса (Arachis) выявило огромное количество неиспользованной генетической изменчивости. Г3 6, 3825–3836. дои: 10.1534/g3.115.026898
, Симпсон С. Э., Чагоя Дж., Мадж Дж. и Буроу М.Д. (2016). Секвенирование транскриптомов различных диких и культивируемых видов арахиса (Arachis) выявило огромное количество неиспользованной генетической изменчивости. Г3 6, 3825–3836. дои: 10.1534/g3.115.026898
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Клевенджер, Дж., Чаварро, К., Перл, С.А., Озиас-Акинс, П., и Джексон, С.А. (2015). Идентификация однонуклеотидного полиморфизма у полиплоидов: обзор, пример и рекомендации. Мол. Завод 8, 831–846.doi: 10.1016/j.molp.2015.02.002
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Клевенджер Дж., Чу Ю., Гимарайнш Л. А., Майя Т., Бертиоли Д., Леал-Бертиоли С. и соавт. (2017). Профилирование экспрессии генов описывает генетическую регуляцию устойчивости Meloidogyne Arenaria в Arachis hypogaea и выявляет ген-кандидат устойчивости. науч. Респ. 7, 1–14.
Академия Google
Датта М. и Редди Л.(1988). Дальнейшие исследования генетики отсутствия клубеньков у арахиса. Растениеводство. 28, 60–62. doi: 10.2135/cropsci1988.0011183x002800010015x
и Редди Л.(1988). Дальнейшие исследования генетики отсутствия клубеньков у арахиса. Растениеводство. 28, 60–62. doi: 10.2135/cropsci1988.0011183x002800010015x
Полнотекстовая перекрестная ссылка | Академия Google
Фу Л., Ню Б., Чжу З., Ву С. и Ли В. (2012). CD-HIT: ускорено для кластеризации данных секвенирования нового поколения. Биоинформатика 28, 3150–3152. doi: 10.1093/биоинформатика/bts565
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Ким, Д., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S.L. (2013). TopHat2: точное выравнивание транскриптомов при наличии вставок, делеций и слияний генов. Геном Биол. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Корани В., Клевенджер Дж. П., Чу Ю. и Озиас-Акинс П. (2019). Машинное обучение как эффективный метод выявления истинных однонуклеотидных полиморфизмов у полиплоидных растений.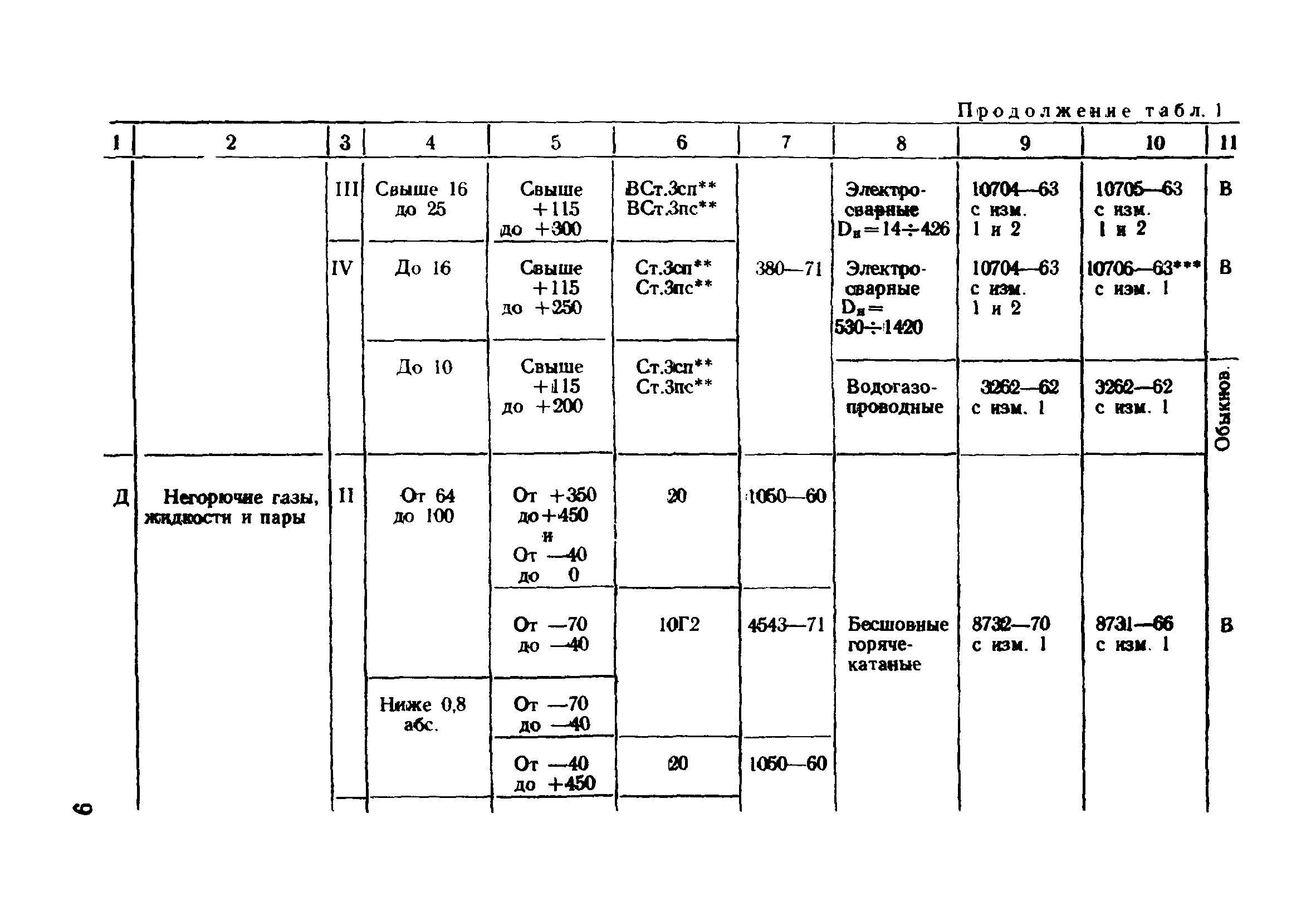 Геном растений 12:180023. doi: 10.3835/plantgenome2018.05.0023
Геном растений 12:180023. doi: 10.3835/plantgenome2018.05.0023
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Ли, Х., Хэндсейкер, Б., Вайсокер, А., Феннелл, Т., Руан, Дж., Гомер, Н., и др. (2009). Формат выравнивания/карты последовательностей и SAMtools. Биоинформатика 25, 2078–2079. doi: 10.1093/биоинформатика/btp352
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Нигам, С., Двиведи, С., и Гиббонс, Р. (1980).«Разведение арахиса в ICRISAT», в материалах Международного семинара по арахису (Патанчеру: Центр ICRISAT).
Академия Google
Олдройд, GE (2013). Говори, дружи и вступай: сигнальные системы, которые способствуют полезным симбиотическим ассоциациям у растений. Нац. Преподобный Микробиолог. 11, 252–263. doi: 10.1038/nrmicro2990
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Панди М.К., Агарвал Г., Кале С. М., Clevenger, J., Nayak, S.N., Sriswathi, M., et al. (2017). Разработка и оценка массива генотипирования высокой плотности «Axiom_Arachis» с 58 тыс. SNP для ускорения генетики и селекции арахиса. науч. Респ. 7:40577. дои: 10.1038/srep40577
М., Clevenger, J., Nayak, S.N., Sriswathi, M., et al. (2017). Разработка и оценка массива генотипирования высокой плотности «Axiom_Arachis» с 58 тыс. SNP для ускорения генетики и селекции арахиса. науч. Респ. 7:40577. дои: 10.1038/srep40577
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Пэн, З., Фан, В., Ван, Л., Паудел, Д., Левентини, Д., Тиллман, Б.Л., и соавт. (2017а). Секвенирование целевого обогащения культивируемого арахиса ( Arachis hypogaea L.) с использованием зондов, сконструированных из последовательностей транскриптов. Мол. Жене. Геномика 292, 955–965. doi: 10.1007/s00438-017-1327-z
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Пэн З., Лю Ф., Ван Л., Чжоу Х., Паудел Д., Тан Л. и др. (2017б). Профили транскриптома показывают генную регуляцию клубеньков арахиса ( Arachis hypogaea L.). науч. Респ. 7:40066. дои: 10.1038/srep40066
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Пэн, З.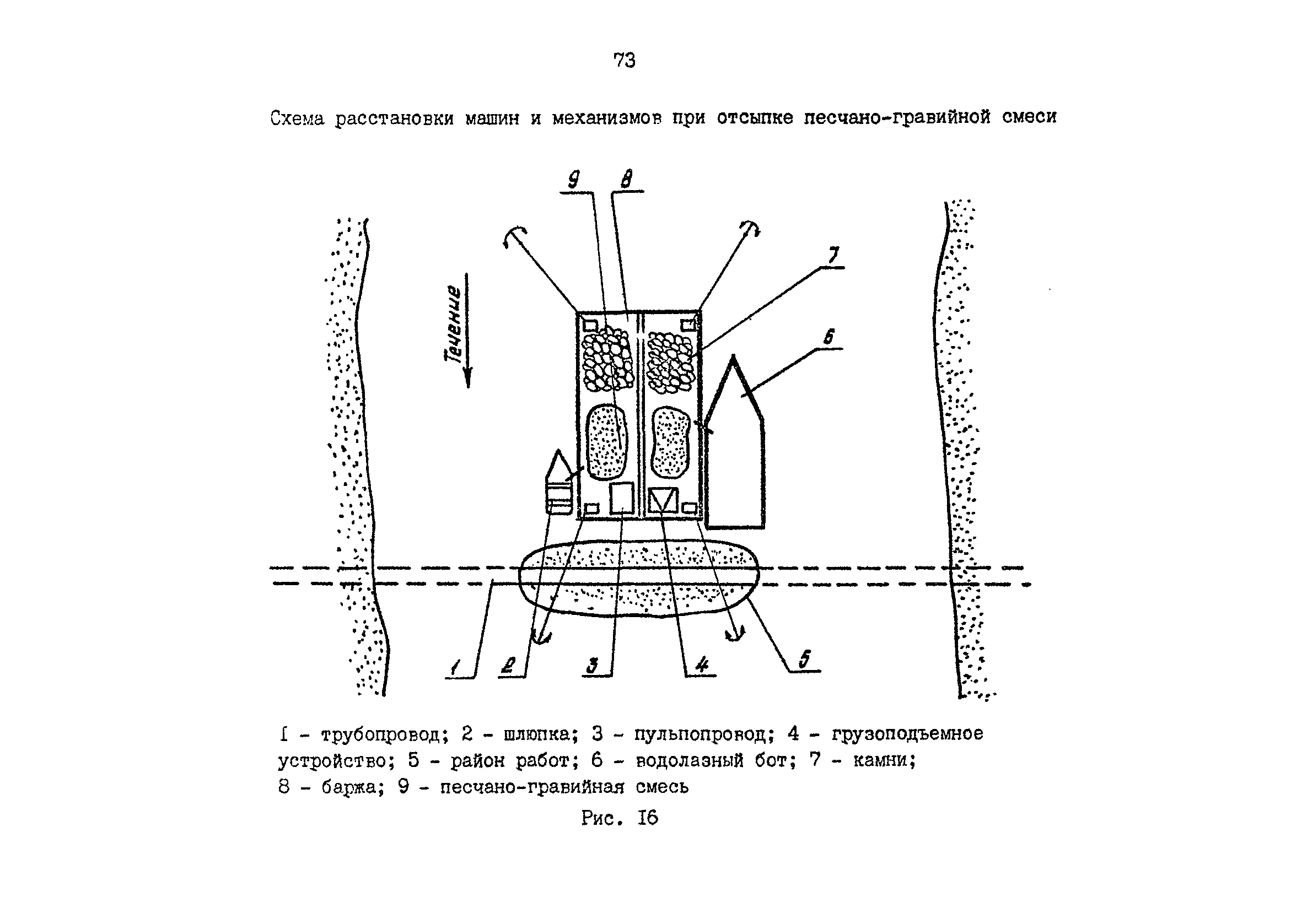 , Tan, L., López, Y., Maku, J., Liu, F., Zhou, H., et al. (2018). Морфологическая и генетическая характеристика неклубеньковых рекомбинантных инбредных линий арахиса. Растениеводство. 58, 540–550. doi: 10.2135/cropsci2017.06.0235
, Tan, L., López, Y., Maku, J., Liu, F., Zhou, H., et al. (2018). Морфологическая и генетическая характеристика неклубеньковых рекомбинантных инбредных линий арахиса. Растениеводство. 58, 540–550. doi: 10.2135/cropsci2017.06.0235
Полнотекстовая перекрестная ссылка | Академия Google
Рен, Б., Ван, X., Дуан, Дж., и Ма, Дж. (2019). Малые РНК, происходящие из ризобиальных тРНК, представляют собой сигнальные молекулы, регулирующие образование клубеньков у растений. Наука 365, 919–922. doi: 10.1126/science.aav8907
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Райс, П., Лонгден, И., и Блисби, А. (2000). EMBOSS: европейский открытый пакет программного обеспечения для молекулярной биологии. Тенденции Жене. 16, 276–277. doi: 10.1016/s0168-9525(00)02024-2
Полнотекстовая перекрестная ссылка | Академия Google
Роджерс С.О. и Бендич А.Дж. (1994). «Извлечение общей клеточной ДНК из растений, водорослей и грибов», в Руководстве по молекулярной биологии растений , под редакцией С.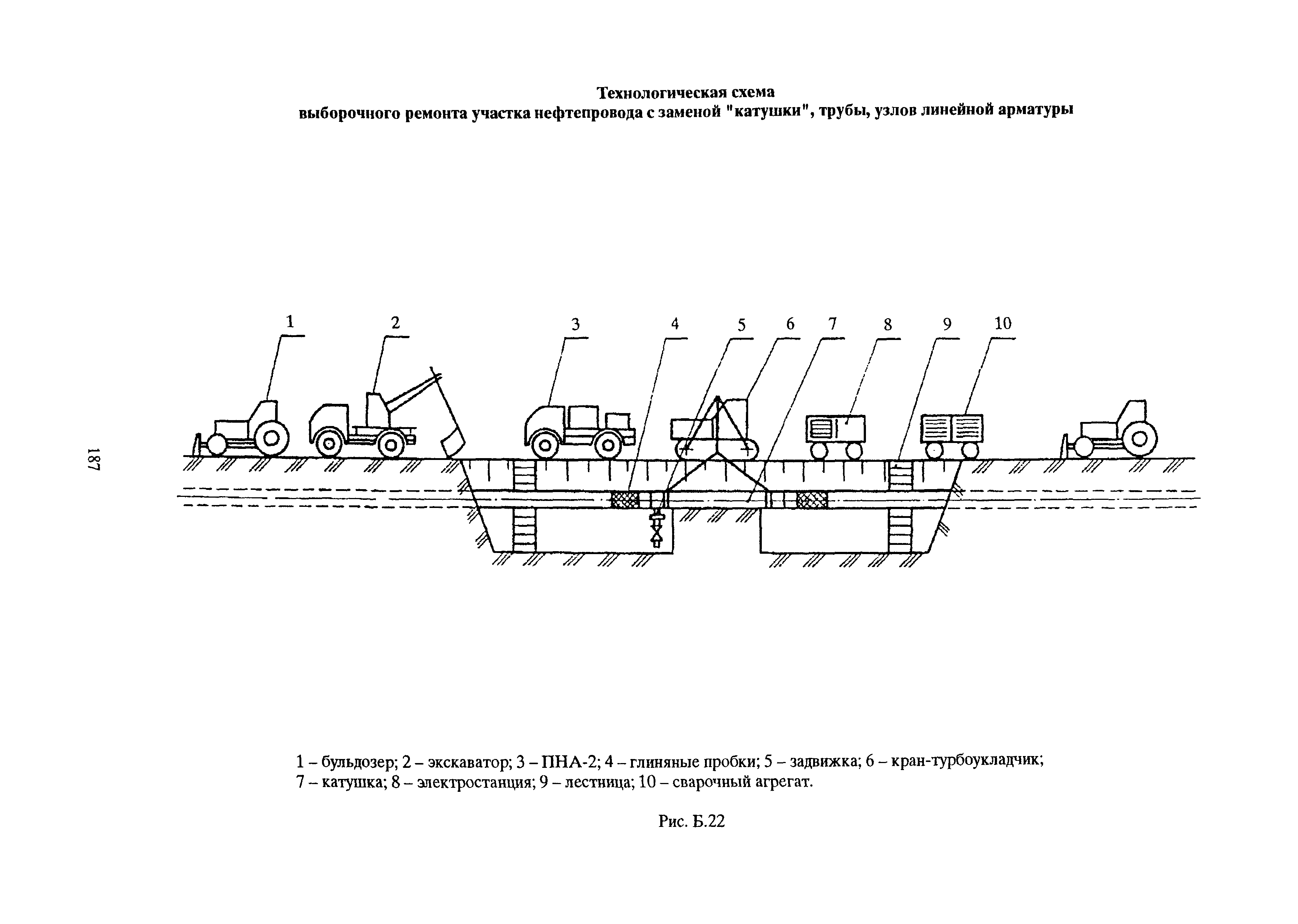 Б. Гельвина и Р. А. Шилпероорта (Дордрехт: Springer), 183–190. дои: 10.1007/978-94-011-0511-8_12
Б. Гельвина и Р. А. Шилпероорта (Дордрехт: Springer), 183–190. дои: 10.1007/978-94-011-0511-8_12
Полнотекстовая перекрестная ссылка | Академия Google
Шварце, К., Бьюкенен, Дж., Тейлор, Дж. К., и Вордсворт, С. (2018). Являются ли подходы полноэкзомного и полногеномного секвенирования рентабельными? Систематический обзор литературы. Жен. Мед. 20, 1122–1130. doi: 10.1038/gim.2017.247
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Ценг Ю., Тиллман Б. Л., Пэн З. и Ван Дж. (2016). Идентификация основных QTL, лежащих в основе устойчивости к вирусу пятнистого увядания томатов, у сорта арахиса Florida-EP TM ‘113’. BMC Genet. 17:128.
Академия Google
Zhou, X., Xia, Y., Ren, X., Chen, Y., Huang, L., Huang, S., et al. (2014). Построение карты генетического сцепления на основе SNP в культивируемом арахисе на основе крупномасштабной разработки маркеров с использованием секвенирования ДНК, связанного с сайтом рестрикции, с двойным перевариванием следующего поколения (ddRADseq).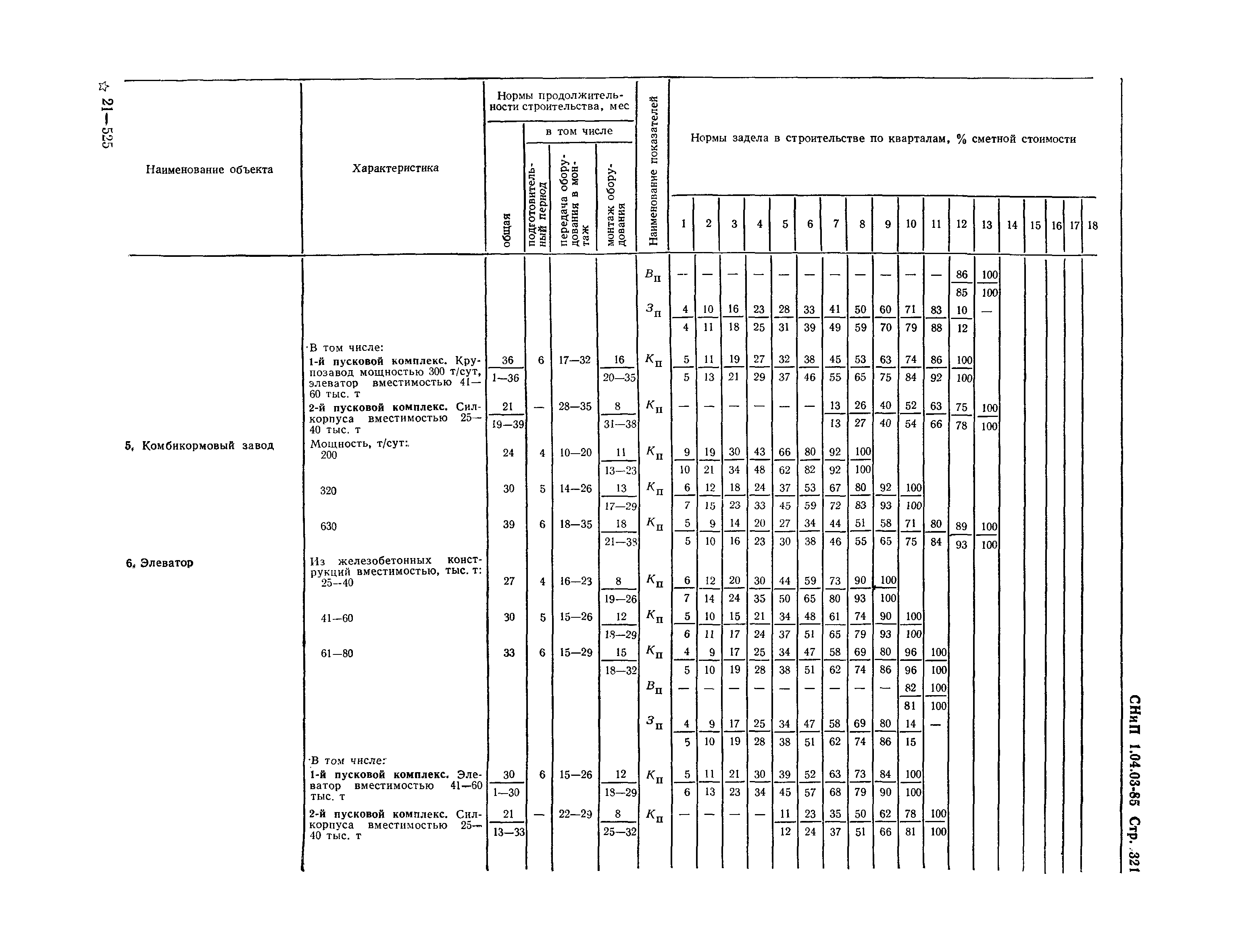 BMC Genomics 15:351. дои: 10.1186/1471-2164-15-351
BMC Genomics 15:351. дои: 10.1186/1471-2164-15-351
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Чжуан В., Чен Х., Yang, M., Wang, J., Pandey, M.K., Zhang, C., et al. (2019). Геном культивируемого арахиса дает представление о кариотипах бобовых, эволюции полиплоидов и одомашнивании сельскохозяйственных культур. Нац. Жене. 51, 865–876. doi: 10.1038/s41588-019-0402-2
Реферат PubMed | Полный текст перекрестной ссылки | Академия Google
Сравнение семи конвейеров и двух технологий секвенирования
Abstract
Секвенирование следующего поколения (NGS) произвело революцию в исследованиях растений и животных во многих отношениях, включая новые методы высокопроизводительного генотипирования.Было продемонстрировано, что генотипирование путем секвенирования (GBS) является надежным и экономически эффективным методом генотипирования, способным давать от тысяч до миллионов SNP для широкого круга видов. Несомненно, самым большим препятствием для его более широкого использования является проблема анализа данных. Здесь мы описываем всестороннее сравнение семи конвейеров биоинформатики GBS, разработанных для обработки необработанных данных о последовательности GBS в генотипы SNP. Мы сравнили пять конвейеров, требующих эталонного генома (TASSEL-GBS v1 и v2, Stacks, IGST и Fast-GBS), и два конвейера de novo , для которых эталонный геном не требуется (UNEAK и Stacks).Используя данные секвенирования Illumina из набора из 24 повторно секвенированных линий сои, мы выполнили вызов SNP с этими конвейерами и сравнили вызовы SNP GBS с данными повторного секвенирования, чтобы оценить их точность. Количество SNP, названных без эталонного генома, было ниже (от 13 000 до 24 000), чем с эталонным геномом (от 25 000 до 54 000 SNP), в то время как точность была высокой (92,3–98,7%) для всех конвейеров, кроме одного (TASSEL-GBSv1, 76,1%). . Среди конвейеров, предлагающих высокую точность (> 95%), Fast-GBS вызвал наибольшее количество полиморфизмов (около 35 000 SNP + Indels) и дал самую высокую точность (98.
Несомненно, самым большим препятствием для его более широкого использования является проблема анализа данных. Здесь мы описываем всестороннее сравнение семи конвейеров биоинформатики GBS, разработанных для обработки необработанных данных о последовательности GBS в генотипы SNP. Мы сравнили пять конвейеров, требующих эталонного генома (TASSEL-GBS v1 и v2, Stacks, IGST и Fast-GBS), и два конвейера de novo , для которых эталонный геном не требуется (UNEAK и Stacks).Используя данные секвенирования Illumina из набора из 24 повторно секвенированных линий сои, мы выполнили вызов SNP с этими конвейерами и сравнили вызовы SNP GBS с данными повторного секвенирования, чтобы оценить их точность. Количество SNP, названных без эталонного генома, было ниже (от 13 000 до 24 000), чем с эталонным геномом (от 25 000 до 54 000 SNP), в то время как точность была высокой (92,3–98,7%) для всех конвейеров, кроме одного (TASSEL-GBSv1, 76,1%). . Среди конвейеров, предлагающих высокую точность (> 95%), Fast-GBS вызвал наибольшее количество полиморфизмов (около 35 000 SNP + Indels) и дал самую высокую точность (98.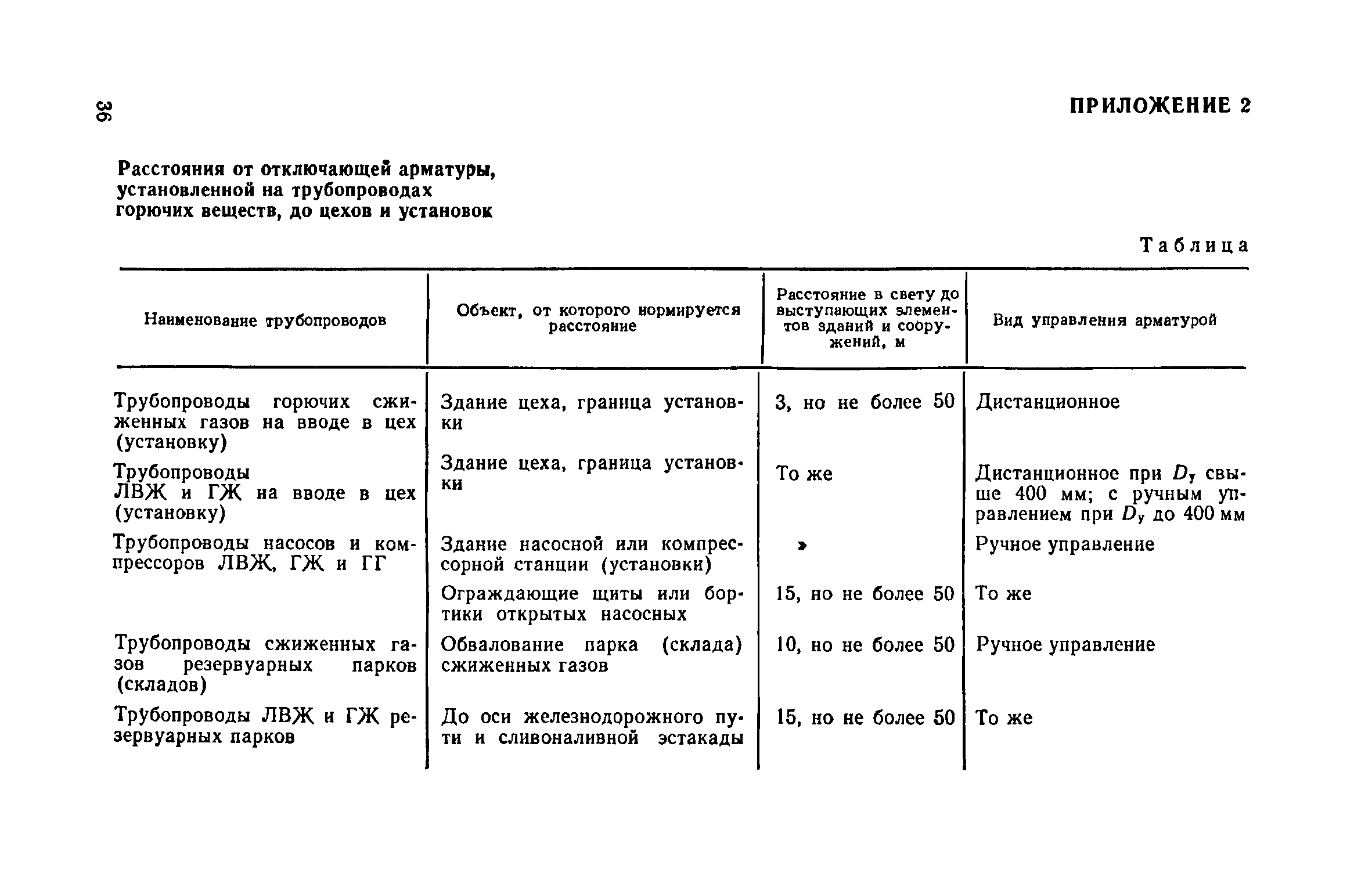 7%). Используя данные последовательности Ion Torrent для тех же 24 строк, мы сравнили производительность Fast-GBS с производительностью TASSEL-GBSv2. Он снова вызвал больше полиморфизмов (25,8 тыс. против 22,9 тыс.), и они оказались более точными (95,2 против 91,1%). Как правило, каталоги SNP, вызываемые из одних и тех же данных секвенирования с использованием разных конвейеров, приводили к сильному перекрытию каталогов SNP (перекрытие 79–92%). Напротив, перекрытие между каталогами SNP, полученными с использованием одного конвейера, но разных технологий секвенирования, было менее значительным (~ 50–70%).
7%). Используя данные последовательности Ion Torrent для тех же 24 строк, мы сравнили производительность Fast-GBS с производительностью TASSEL-GBSv2. Он снова вызвал больше полиморфизмов (25,8 тыс. против 22,9 тыс.), и они оказались более точными (95,2 против 91,1%). Как правило, каталоги SNP, вызываемые из одних и тех же данных секвенирования с использованием разных конвейеров, приводили к сильному перекрытию каталогов SNP (перекрытие 79–92%). Напротив, перекрытие между каталогами SNP, полученными с использованием одного конвейера, но разных технологий секвенирования, было менее значительным (~ 50–70%).
Введение
Секвенирование следующего поколения (NGS) значительно облегчило разработку методов генотипирования очень большого числа молекулярных маркеров, таких как однонуклеотидные полиморфизмы (SNP). NGS предлагает несколько подходов, которые способны одновременно выполнять обнаружение SNP по всему геному и генотипирование за один шаг, даже у видов, для которых мало или вообще нет генетической информации [1].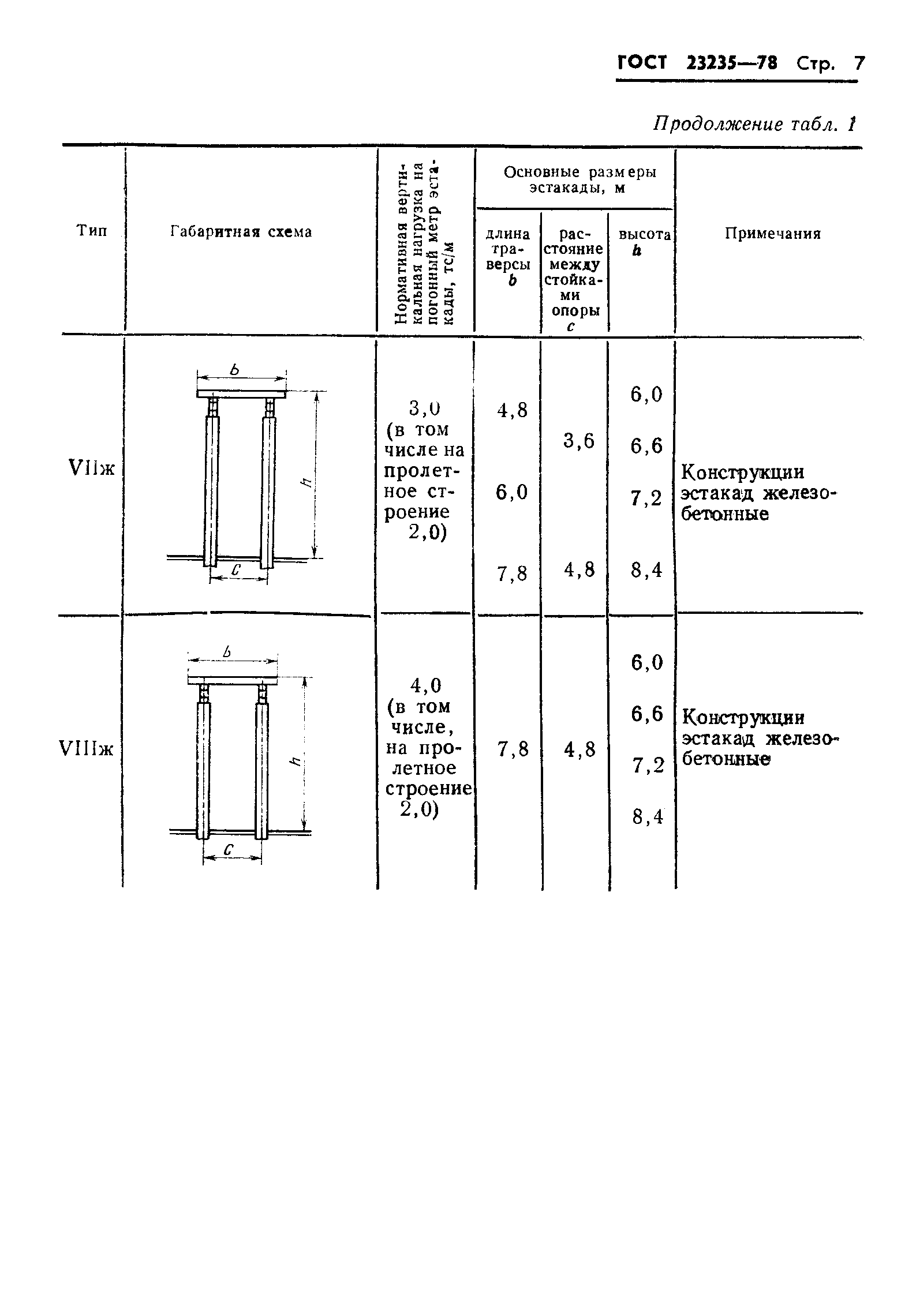 Эта революция в открытии генетических маркеров позволяет изучать важные вопросы молекулярной селекции, популяционной генетики, экологической генетики и эволюции.Наиболее широко используемые методы генотипирования, основанные на NGS, используют рестрикционные ферменты для захвата уменьшенного представления генома [2–9]. Новые подходы, такие как секвенирование ДНК, связанное с сайтом рестрикции (RAD-seq) и генотипирование посредством секвенирования (GBS), были разработаны в качестве быстрых и надежных подходов к секвенированию мультиплексированных образцов с уменьшенным представлением, которое сочетает в себе обнаружение молекулярных маркеров по всему геному и генотипирование. [1]. Это семейство подходов к генотипированию с уменьшенным представлением, обычно называемое генотипированием путем секвенирования (GBS) [1].Гибкость и низкая стоимость GBS делают его отличным инструментом для многих приложений и исследовательских вопросов в области генетики и селекции. Такие современные достижения позволяют генотипировать тысячи SNP, при этом вероятность выявления SNP, коррелирующих с интересующими признаками, возрастает [10].
Эта революция в открытии генетических маркеров позволяет изучать важные вопросы молекулярной селекции, популяционной генетики, экологической генетики и эволюции.Наиболее широко используемые методы генотипирования, основанные на NGS, используют рестрикционные ферменты для захвата уменьшенного представления генома [2–9]. Новые подходы, такие как секвенирование ДНК, связанное с сайтом рестрикции (RAD-seq) и генотипирование посредством секвенирования (GBS), были разработаны в качестве быстрых и надежных подходов к секвенированию мультиплексированных образцов с уменьшенным представлением, которое сочетает в себе обнаружение молекулярных маркеров по всему геному и генотипирование. [1]. Это семейство подходов к генотипированию с уменьшенным представлением, обычно называемое генотипированием путем секвенирования (GBS) [1].Гибкость и низкая стоимость GBS делают его отличным инструментом для многих приложений и исследовательских вопросов в области генетики и селекции. Такие современные достижения позволяют генотипировать тысячи SNP, при этом вероятность выявления SNP, коррелирующих с интересующими признаками, возрастает [10]. Даже с продвижением NGS для получения миллионов считываний последовательностей за цикл анализ данных для этих новых подходов может быть сложным из-за использования рестрикционных ферментов, мультиплексирования образцов, различной длины фрагмента и переменной глубины считывания [1].Совершенно очевидно, что конвейеры расширенного анализа стали необходимостью для фильтрации, сортировки и выравнивания этих данных последовательности. Конвейер для GBS должен включать шаги для фильтрации некачественных прочтений, классификации прочтений по пулу или отдельным лицам на основе штрих-кодов последовательности, либо идентификации локусов и аллелей de novo , либо сопоставления прочтений с эталонным геномом индекса для обнаружения полиморфизмов и часто оценка генотипы для каждой особи, включенной в исследование. Как правило, конвейеры для обработки данных GBS делятся на две группы; de novo на основе и на основе ссылок.Когда эталонный геном доступен, чтения из секвенирования с уменьшенным представлением могут быть сопоставлены с эталонным геномом, а SNP могут быть вызваны как для проектов ресеквенирования всего генома [11-12].
Даже с продвижением NGS для получения миллионов считываний последовательностей за цикл анализ данных для этих новых подходов может быть сложным из-за использования рестрикционных ферментов, мультиплексирования образцов, различной длины фрагмента и переменной глубины считывания [1].Совершенно очевидно, что конвейеры расширенного анализа стали необходимостью для фильтрации, сортировки и выравнивания этих данных последовательности. Конвейер для GBS должен включать шаги для фильтрации некачественных прочтений, классификации прочтений по пулу или отдельным лицам на основе штрих-кодов последовательности, либо идентификации локусов и аллелей de novo , либо сопоставления прочтений с эталонным геномом индекса для обнаружения полиморфизмов и часто оценка генотипы для каждой особи, включенной в исследование. Как правило, конвейеры для обработки данных GBS делятся на две группы; de novo на основе и на основе ссылок.Когда эталонный геном доступен, чтения из секвенирования с уменьшенным представлением могут быть сопоставлены с эталонным геномом, а SNP могут быть вызваны как для проектов ресеквенирования всего генома [11-12]. К настоящему времени было разработано несколько конвейеров анализа GBS на основе эталонов. Наиболее широко используемыми конвейерами анализа GBS на основе эталонов являются: TASSEL-GBS (v1 и v2), Stacks, IGST и Fast-GBS (самый последний конвейер, Torkamaneh et al. (неопубликованный)) [9, 13–15]. . В отсутствие эталонного генома необходимо идентифицировать пары почти идентичных прочтений (предположительно представляющих альтернативные аллели локуса).Наиболее часто используемыми конвейерами для такого подхода на основе de novo являются UNEAK и Stacks [15, 16].
К настоящему времени было разработано несколько конвейеров анализа GBS на основе эталонов. Наиболее широко используемыми конвейерами анализа GBS на основе эталонов являются: TASSEL-GBS (v1 и v2), Stacks, IGST и Fast-GBS (самый последний конвейер, Torkamaneh et al. (неопубликованный)) [9, 13–15]. . В отсутствие эталонного генома необходимо идентифицировать пары почти идентичных прочтений (предположительно представляющих альтернативные аллели локуса).Наиболее часто используемыми конвейерами для такого подхода на основе de novo являются UNEAK и Stacks [15, 16].
Наконец, в настоящее время доступны различные платформы секвенирования NGS, предлагающие различные преимущества. Например, в то время как технология Illumina предлагает очень высокую пропускную способность и качество чтения, это обычно достигается за счет скорости, поскольку для завершения цикла требуется около двух недель. Напротив, технология Ion Torrent [17] предлагает большую скорость (4 часа) за счет более низкой пропускной способности и качества чтения. В зависимости от ограничений та или иная технология может оказаться более подходящей. В идеале хотелось бы, чтобы конвейеры вызовов SNP одинаково хорошо работали с обоими типами считываемых данных.
В зависимости от ограничений та или иная технология может оказаться более подходящей. В идеале хотелось бы, чтобы конвейеры вызовов SNP одинаково хорошо работали с обоими типами считываемых данных.
В этом исследовании мы всесторонне сравнили существующие конвейеры анализа GBS на основе количества вызванных SNP, точности полученных генотипов, а также скорости и простоты использования этих конвейеров. Мы также сравнили результаты, полученные с помощью ридов Illumina и Ion Torrent. Наконец, мы изучили количество перекрытий в локусах SNP, которые вызывались с использованием разных конвейеров.
Материалы и методы
Образцы и платформа для секвенирования
Соя ( Glycine max L .) представляет собой диплоидный вид с 20 парами хромосом и геномом среднего размера (1,1 Гб). Поскольку это автогамный вид, линии/сорта сои размножаются правильно и в высокой степени гомозиготны. Набор из 23 канадских линий сои и одной интродукции растений (PI) был подвергнут анализу GBS. Эти же линии были повторно секвенированы, как описано ранее Torkamaneh и Belzile [18].Используя одну и ту же ДНК, две библиотеки GBS были созданы после расщепления Ape KI: одна для секвенирования Illumina (согласно Elshire et al. [6]), а другая для секвенирования Ion Torrent (согласно Mascher et al. [19]). . Одностороннее секвенирование проводили либо на Illumina HiSeq 2000 в Инновационном центре McGill University-Génome Québec в Монреале, Канада, либо на машине Ion Proton в Институте интегративной и системной биологии (IBIS) Университета Лаваля, Квебек. Канада. Всего на платформе Illumina было сгенерировано 42 миллиона прочтений размером 100 п.н., а на платформе Ion Torrent — 38 миллионов прочтений размером от 50 до 135 п.н.Все данные (GBS и WGS) доступны в архиве NCBI Sequence Read Archive (SRA) под номерами SRP059747 (последовательности Illumina) и SRP073237 (последовательности Ion Torrent).
Эти же линии были повторно секвенированы, как описано ранее Torkamaneh и Belzile [18].Используя одну и ту же ДНК, две библиотеки GBS были созданы после расщепления Ape KI: одна для секвенирования Illumina (согласно Elshire et al. [6]), а другая для секвенирования Ion Torrent (согласно Mascher et al. [19]). . Одностороннее секвенирование проводили либо на Illumina HiSeq 2000 в Инновационном центре McGill University-Génome Québec в Монреале, Канада, либо на машине Ion Proton в Институте интегративной и системной биологии (IBIS) Университета Лаваля, Квебек. Канада. Всего на платформе Illumina было сгенерировано 42 миллиона прочтений размером 100 п.н., а на платформе Ion Torrent — 38 миллионов прочтений размером от 50 до 135 п.н.Все данные (GBS и WGS) доступны в архиве NCBI Sequence Read Archive (SRA) под номерами SRP059747 (последовательности Illumina) и SRP073237 (последовательности Ion Torrent).
Конвейеры анализа GBS
Мы использовали два вызывающих варианта de novo и пять эталонных конвейеров (эталонный геном Williams82; [20]) для вызова SNP. Мы запускали все пайплайны в одинаковых условиях глубины охвата (minDP≥2), максимального несоответствия для выравнивания (n = 3), максимального количества отсутствующих данных (MaxMD = 80%) и минимальной частоты минорных аллелей (MinMAF≥0.05). Ниже мы кратко опишем процессы для каждого пайплайна. Для вычислений мы использовали систему Linux с 10 ЦП и 25 ГБ памяти. В дополнение к описаниям, представленным ниже, в таблице S1 приведена сводка различных компонентов каждого конвейера, и мы предоставляем все командные строки, используемые в этой работе, в качестве вспомогательной информации (текст S1).
Мы запускали все пайплайны в одинаковых условиях глубины охвата (minDP≥2), максимального несоответствия для выравнивания (n = 3), максимального количества отсутствующих данных (MaxMD = 80%) и минимальной частоты минорных аллелей (MinMAF≥0.05). Ниже мы кратко опишем процессы для каждого пайплайна. Для вычислений мы использовали систему Linux с 10 ЦП и 25 ГБ памяти. В дополнение к описаниям, представленным ниже, в таблице S1 приведена сводка различных компонентов каждого конвейера, и мы предоставляем все командные строки, используемые в этой работе, в качестве вспомогательной информации (текст S1).
Fast-GBS
Конвейер анализа Fast-GBS был разработан путем интеграции общедоступных пакетов с инструментами собственной разработки. Основные функции включают в себя: (1) демультиплексирование и очистку считываний необработанных последовательностей; (2) оценка качества чтения и составление карт; (3) фильтрация сопоставленных прочтений и оценка сложности библиотеки; (4) перегруппировка и построение локальных гаплотипов; (5) подходящие популяционные частоты и отдельные гаплотипы; (5) необработанный вариант вызова; (6) вариантная и индивидуальная фильтрация; (7) идентификация высококонсистентных вариантов. Поскольку исследователи не всегда могут иметь немедленный доступ к ресурсам кластера, этот конвейер позволяет либо параллельную обработку большого количества образцов в кластере, либо последовательную обработку нескольких образцов на одной машине.
Поскольку исследователи не всегда могут иметь немедленный доступ к ресурсам кластера, этот конвейер позволяет либо параллельную обработку большого количества образцов в кластере, либо последовательную обработку нескольких образцов на одной машине.
IGST (инструмент IBIS для генотипирования посредством секвенирования)
Конвейер, реализованный на языке программирования Perl, был разработан для обработки данных считывания последовательностей Illumina. Шаги, задействованные в конвейере, выполнялись в отдельных сценариях оболочки. В этом конвейере используются различные общедоступные программные инструменты (набор инструментов FASTX, BWA, SAMtools, VCFtools), а также некоторые собственные инструменты [11, 21, 22].Полученные необработанные SNP были дополнительно отфильтрованы с использованием VCFtools на основе глубины чтения, отсутствующих данных в генотипах и частоте минорных аллелей. Гетерозиготная коррекция выполняется собственным скриптом Python.
TASSEL-GBS (версии 1 и 2)
Конвейеры TASSEL-GBS реализованы на языке программирования Java. В настоящее время доступны две версии: TASSEL-GBS v1 (TASSEL 3.0) [13] и TASSEL-GBS v2 (TASSEL 5.0) [14]. Оба конвейера работают одинаково и требуют, чтобы все операции чтения были урезаны до одинаковой длины (64 п.н. в версии 1, до 92 п.н. в версии 2), а идентичные операции чтения свернуты в теги.Эти теги затем выравниваются по отношению к эталонному геному, и из выровненных тегов вызываются SNP. Основными изменениями, реализованными в TASSEL-GBS v2, являются: 1) возможность использования более длинных меток для повышения точности выравнивания с эталонным геномом и 2) расширенный этап обнаружения и производства SNP.
В настоящее время доступны две версии: TASSEL-GBS v1 (TASSEL 3.0) [13] и TASSEL-GBS v2 (TASSEL 5.0) [14]. Оба конвейера работают одинаково и требуют, чтобы все операции чтения были урезаны до одинаковой длины (64 п.н. в версии 1, до 92 п.н. в версии 2), а идентичные операции чтения свернуты в теги.Эти теги затем выравниваются по отношению к эталонному геному, и из выровненных тегов вызываются SNP. Основными изменениями, реализованными в TASSEL-GBS v2, являются: 1) возможность использования более длинных меток для повышения точности выравнивания с эталонным геномом и 2) расширенный этап обнаружения и производства SNP.
UNEAK (Универсальный набор для анализа с поддержкой сети)
Общий дизайн UNEAK следующий: 1) считывания обрезаются до 64 п.н.; 2) идентичные риды длиной 64 п.н. свернуты в теги; 3) попарное выравнивание идентифицирует пары тегов, имеющие несовпадение одной пары оснований.Эти несовпадения в одной паре оснований являются кандидатами в SNP. «Сетевой фильтр» используется для отбрасывания повторов, паралогов и ошибок секвенирования, что приводит к набору взаимных пар тегов или SNP.
Стеки (на основе эталона и
de novo ) Необработанные входные данные для стеков представляют собой секвенированные фрагменты ДНК из любого протокола GBS на основе рестрикционных ферментов. Стеки могут обрабатывать необработанные данные секвенирования для идентификации локусов de novo или путем выравнивания с эталонным геномом [10].Независимо от того, собраны ли данные de novo или сопоставлены с эталонным геномом, многие последующие шаги в стеках являются общими. Конвейер можно описать следующим образом: (1) Необработанные чтения последовательностей демультиплексируются и очищаются (process_radtags). (2) Данные от каждого индивидуума группируются в локусы и идентифицируются сайты полиморфных нуклеотидов (ustacks или pstacks для невыровненных или выровненных данных соответственно). (3) Локусы группируются по отдельным лицам, и составляется каталог (cstacks).(4) Локусы от каждого индивидуума сопоставляются с каталогом для определения аллельного состояния в каждом локусе у каждого индивидуума (sstacks). (5) Аллельные состояния либо преобразуются в набор картируемых генотипов (для генетической карты) с использованием генотипов, либо подвергаются популяционно-генетической статистике через популяции с записью результатов в один или несколько выходных файлов.
(5) Аллельные состояния либо преобразуются в набор картируемых генотипов (для генетической карты) с использованием генотипов, либо подвергаются популяционно-генетической статистике через популяции с записью результатов в один или несколько выходных файлов.
Точность генотипа
Для оценки точности названий генотипов мы использовали собственный сценарий для сравнения генотипов, названных с использованием GBS, с генотипами, названными в тех же локусах после WGS.Секвенирование и определение SNP в этой коллекции из 24 линий сои ранее было описано Torkamaneh и Belzile [18]. Вкратце, линии сои были секвенированы до средней глубины охвата 9x, и было достигнуто покрытие генома 96%. Чтения парных концов Illumina были сопоставлены с эталонным геномом сои (Williams82) с использованием BWA, а генотипы в полиморфных локусах были названы с помощью SAMtools. Варианты с двумя или более альтернативными аллелями были удалены. Таким образом, среди этих линий было названо в общей сложности 3,6 млн SNP.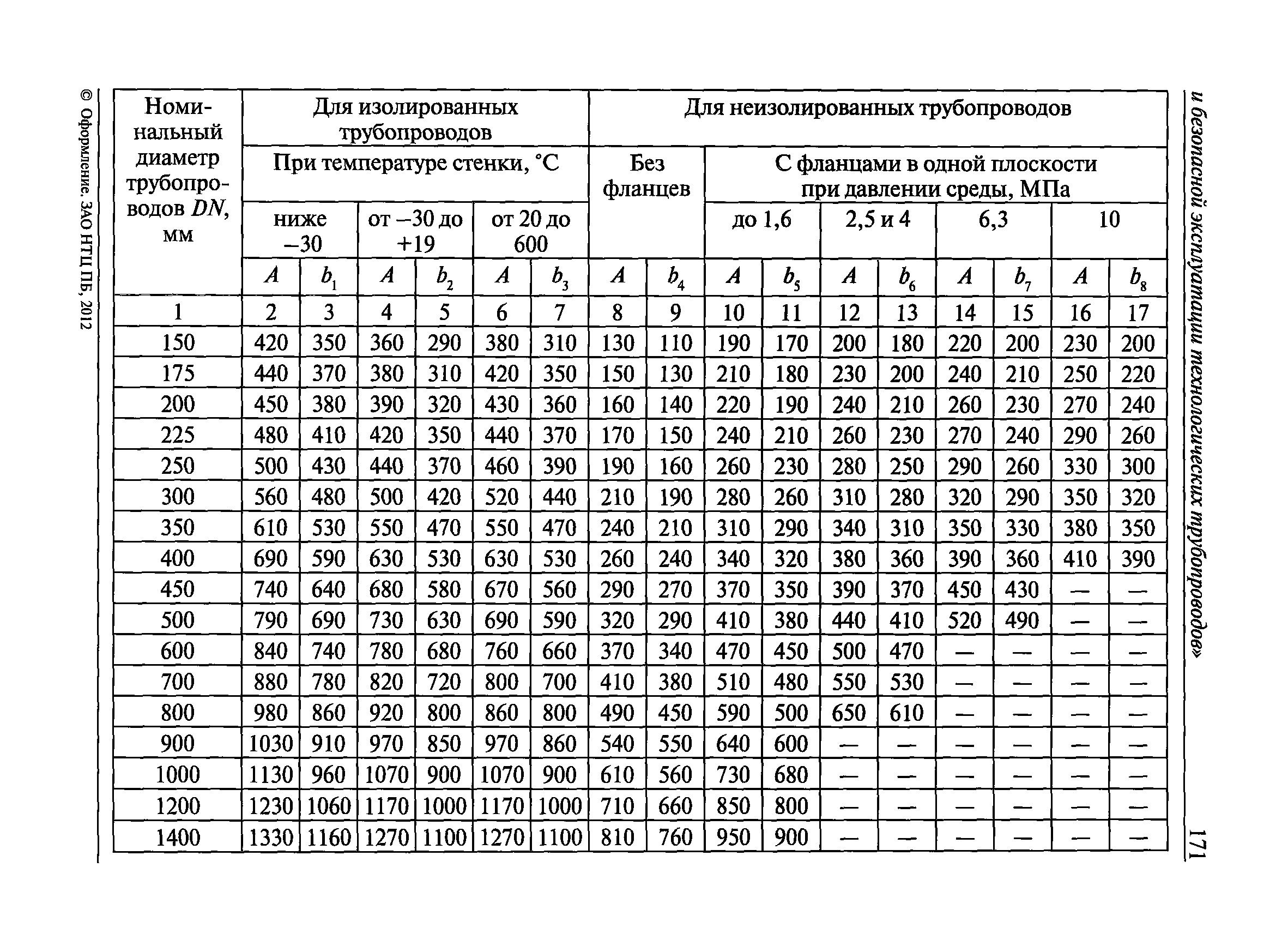 В качестве дополнительного средства для измерения качества генотипа мы оценили долю отсутствующих данных и гетерозиготных вызовов, полученных с каждым конвейером анализа. Для конвейеров de novo мы сопоставили теги, поддерживающие SNP, с эталонным геномом, чтобы найти физическое положение, а затем сравнили их с набором данных WGS.
В качестве дополнительного средства для измерения качества генотипа мы оценили долю отсутствующих данных и гетерозиготных вызовов, полученных с каждым конвейером анализа. Для конвейеров de novo мы сопоставили теги, поддерживающие SNP, с эталонным геномом, чтобы найти физическое положение, а затем сравнили их с набором данных WGS.
Результаты
Вызов вариантов с различными конвейерами с использованием данных чтения Illumina
Чтобы оценить эффективность различных конвейеров анализа GBS, мы проанализировали общедоступные данные GBS (чтения Illumina 100 п.н.) из набора из 24 ранее изученных линий сои.Мы сравнили пять эталонных конвейеров анализа: TASSEL-GBS v1 и v2, Stacks, IGST и Fast-GBS. Мы также сравнили два широко используемых вызывающих абонента de novo : UNEAK и Stacks. Мы использовали одинаковое количество чтений для всех анализов (42M чтений) и попытались подобрать параметры, максимально схожие для всех пайплайнов (подробности см. в M&M). Как показано на рисунке, большие различия в количестве вызванных SNP наблюдались как для de novo , так и для эталонных конвейеров.Среди первых Стэкс назвал наименьшее количество SNP, примерно в 2 раза меньше, чем UNEAK (13 303 против 24 743). Количество SNP, вызванных UNEAK, было ненамного ниже среднего количества SNP, вызванных референсными конвейерами (32 423). Среди эталонных конвейеров количество вызванных SNP варьировалось от 18 941 (стеки) до 54 412 (TASSEL-GBS v1), т. е. разница в 2,8 раза. Три других референсных пайплайна были намного ближе к среднему значению, вызывая примерно от 25 до 35 тысяч SNP. Помимо вызова SNP, IGST и Fast-GBS также могли вызывать вставки.В обоих случаях они внесли дополнительные 12–13% в общее количество вариантов.
Как показано на рисунке, большие различия в количестве вызванных SNP наблюдались как для de novo , так и для эталонных конвейеров.Среди первых Стэкс назвал наименьшее количество SNP, примерно в 2 раза меньше, чем UNEAK (13 303 против 24 743). Количество SNP, вызванных UNEAK, было ненамного ниже среднего количества SNP, вызванных референсными конвейерами (32 423). Среди эталонных конвейеров количество вызванных SNP варьировалось от 18 941 (стеки) до 54 412 (TASSEL-GBS v1), т. е. разница в 2,8 раза. Три других референсных пайплайна были намного ближе к среднему значению, вызывая примерно от 25 до 35 тысяч SNP. Помимо вызова SNP, IGST и Fast-GBS также могли вызывать вставки.В обоих случаях они внесли дополнительные 12–13% в общее количество вариантов.
Таблица 1
Количество SNP и вставок, обнаруженных среди 24 линий сои с использованием семи различных конвейеров биоинформатики в считываниях Illumina. Также предоставляются время и объем памяти, необходимые для запуска каждого конвейера.
| Варианты | |||||
|---|---|---|---|---|---|
| подход | Pipeline | ОНП | вставкам | Время * (час: мин) | Память (Gb) |
| Стеки 13303 | ND 3:07 | ||||
| UNEAK 24743 | ND 1:11 | 20 | |||
| Справочно на основе | кисточкой-GBSv1 | 54,412 ND | 1:45 15 | ||
| Стеки 18941 | ND 3:30 | 14 | |||
| IGST 25650 | 3,170 | 12:59 | 240 | ||
| TASSEL-GBSv2 | 28,158 | ND | 4:16 | 1 18 | |
| Fast-GBS | 34 953 | 3 921 | 1:47 | 1:47 | 27 |
Fast-GBS и Tassel-GBS V1 оказались самым быстрым бегом среди опорных трубопроводов (~ 1H55 ), тогда как IGST оказался самым медленным, для завершения анализа потребовалось почти 13 часов. Среди конвейеров de novo UNEAK был почти в три раза быстрее, чем Stacks (1 ч 21 мин против 3 ч 07 мин) и оказался самым быстрым из всех конвейеров. С точки зрения требуемой памяти здесь также наблюдались очень большие различия. Среди конвейеров de novo для UNEAK требовалось почти в три раза больше места на диске по сравнению со стеками (20 Гб против 7 Гб). Среди эталонных конвейеров различия были еще больше, поскольку для IGST требовалось в 17,1 раза больше памяти (240 Гб), чем для стеков (14 Гб).
Среди конвейеров de novo UNEAK был почти в три раза быстрее, чем Stacks (1 ч 21 мин против 3 ч 07 мин) и оказался самым быстрым из всех конвейеров. С точки зрения требуемой памяти здесь также наблюдались очень большие различия. Среди конвейеров de novo для UNEAK требовалось почти в три раза больше места на диске по сравнению со стеками (20 Гб против 7 Гб). Среди эталонных конвейеров различия были еще больше, поскольку для IGST требовалось в 17,1 раза больше памяти (240 Гб), чем для стеков (14 Гб).
Точность и эффективность конвейеров биоинформатики GBS
Чтобы изучить качество данных SNP, полученных с использованием эталонных конвейеров, мы сначала измерили количество недостающих данных, а затем оценили точность генотипа, сравнив генотипы, полученные из GBS, с истинными генотипами. обнаружены с помощью полногеномного повторного секвенирования тех же линий.Оценки точности SNP, называемых GBS, были выполнены для всех SNP для всех пайплайнов при одинаковых уровнях допуска отсутствующих данных (≤80%) и частоте минорных аллелей (≥0,05). Как видно из , среди эталонных пайплайнов доля отсутствующих данных варьировалась от 28 % (TASSEL GBS v1) до 57,3 % (стеки). Среди конвейеров de novo доля отсутствующих данных была менее изменчивой: от 39,4% (стеки) до 41,3% (UNEAK).
Как видно из , среди эталонных пайплайнов доля отсутствующих данных варьировалась от 28 % (TASSEL GBS v1) до 57,3 % (стеки). Среди конвейеров de novo доля отсутствующих данных была менее изменчивой: от 39,4% (стеки) до 41,3% (UNEAK).
Таблица 2
Точность данных GBS SNP, полученных с платформы Illumina с использованием другого конвейера биоинформатики.
| Подход | De NOVO | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Параметр / трубопровод | Стеки | Tassel-GBS V1 | Tassel-GBS V1 | Стеки | IGST | Tassel-GBS V2 | Fast-GBS | |||||||
| Количество ОНП | 13303 | 24743 | 54412 | 18941 | 25650 | 28158 | 34953 | |||||||
| Количество генотипов | 319272 | 593832 | 1305888 41.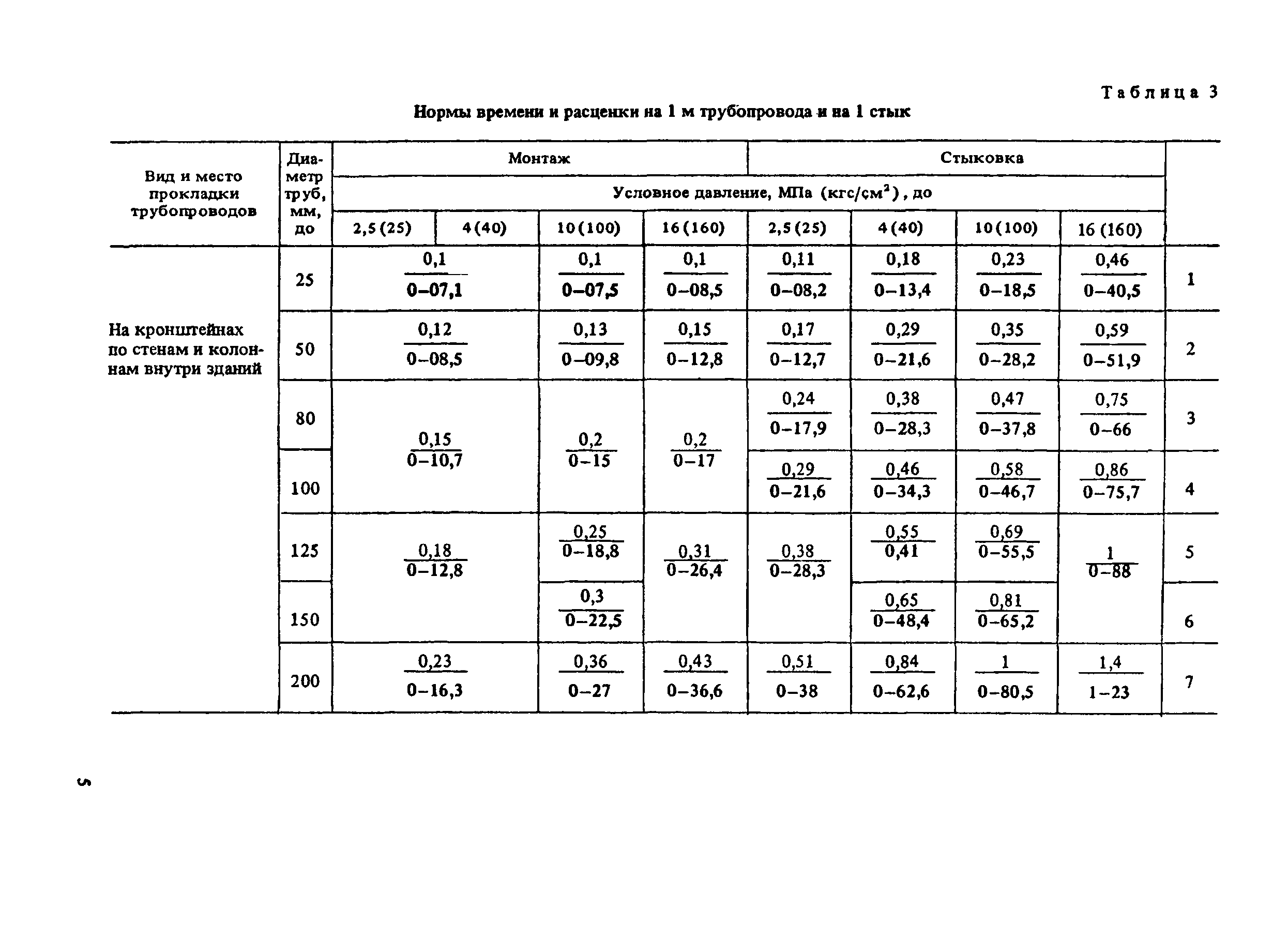 3 3 | 39,4 | 28 | 57,3 | 44 | 35,6 | 46 | |||||
| гетерозигот (%) | 3,7 | 5,3 | 11,5 | 4,4 | 5,9 | 5,7 | 3,4 | |||||||
| Loci с> 50% гетерозиготы * | 0 | 0 | 65 | 65 | 324 | 551 | 551 | 184 | ||||||
| Точность (%) | 93.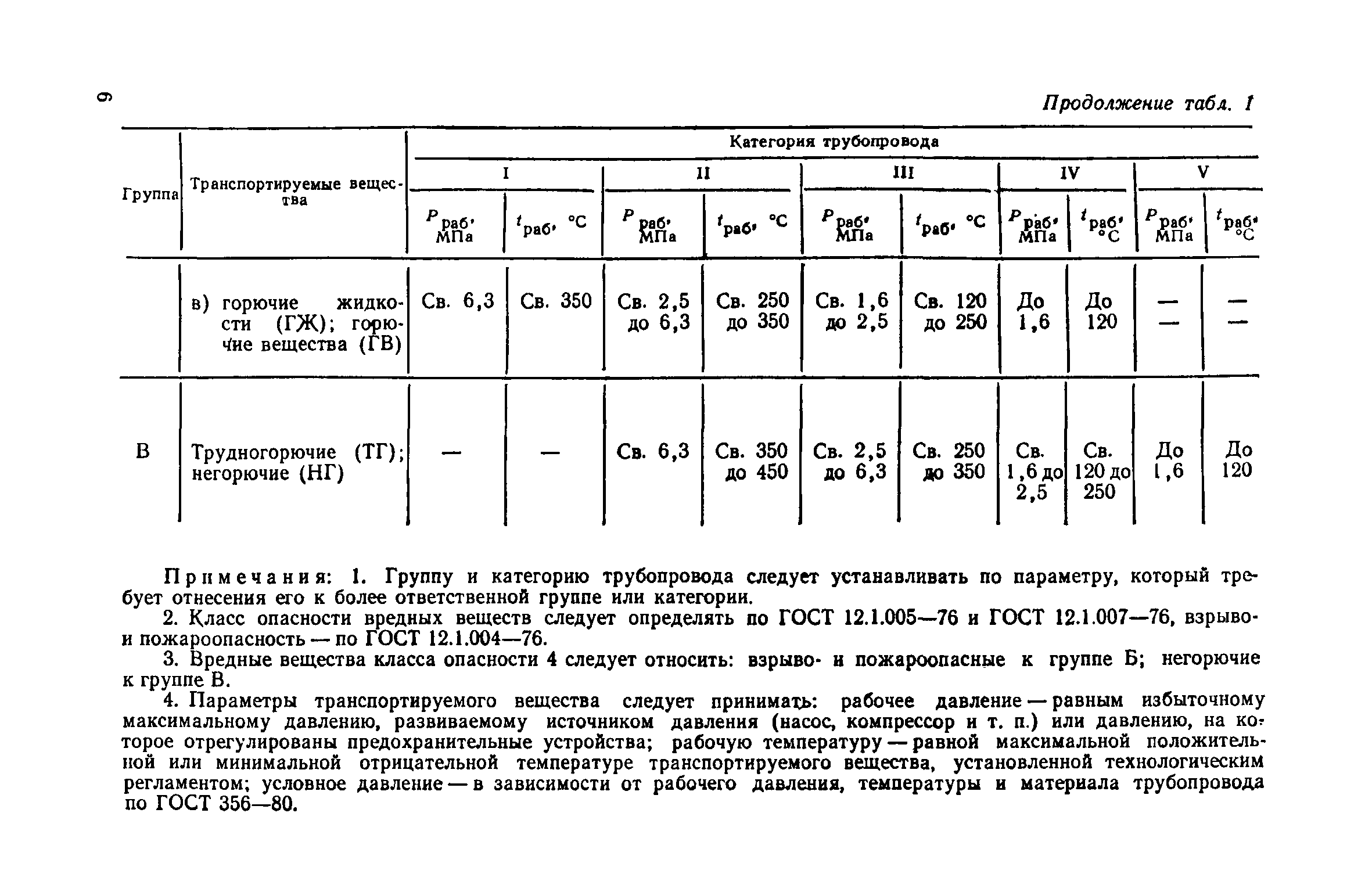 6 6 | 93872 | 93.9 | 76.1 | 93.2 | 93.2 | 984 | 98.4 | 92.3 | 92.3 | 98.9 | 98,7 | 98,7 | 98,760 |
7
, когда мы сравниваем генотипы, полученные с использованием каждого трубопровода с генотипами, полученными из резюме, мы обнаружили, что 98,7% генотипов SNP под названием конвейер Fast-GBS соответствовал истинным генотипам. Подобные уровни точности были обнаружены для SNP, вызванных с помощью IGST (98,4%). За единственным исключением, все эталонные пайплайны достигли уровня точности >92%.TASSEL-GBS v1 оказался наименее точным из этих конвейеров, поскольку только 76,1% названных им генотипов были идентичны данным повторного секвенирования. Среди конвейеров de novo точность вызовов генотипа была лишь немного ниже (в среднем 93,7%), чем точность, полученная с эталонными конвейерами, отличными от TASSEL-GBS v1 (в среднем 95,6%).
За единственным исключением, все эталонные пайплайны достигли уровня точности >92%.TASSEL-GBS v1 оказался наименее точным из этих конвейеров, поскольку только 76,1% названных им генотипов были идентичны данным повторного секвенирования. Среди конвейеров de novo точность вызовов генотипа была лишь немного ниже (в среднем 93,7%), чем точность, полученная с эталонными конвейерами, отличными от TASSEL-GBS v1 (в среднем 95,6%).
Среди растений недавние или древние события полиплоидизации могут генерировать паралоги, которые могут быть ошибочно приняты за аллели одного локуса на основании чтения коротких последовательностей.Поэтому мы исследовали как общее количество гетерозиготных вызовов генотипа, так и количество локусов, содержащих большую долю (> 50%) гетерозиготных вызовов. Как видно из , de novo конвейеров вызвали аналогичную долю гетерозиготных генотипов (~ 3,7 и 5,3% для Stacks и UNEAK соответственно) и не сохранили ни одного локуса с большой долей гетерозигот. Среди эталонных конвейеров Fast-GBS и TASSEL-GBS v1 назвали наименьшее количество и самые гетерозиготные генотипы (3.4 и 11,5% соответственно). Кроме того, TASSEL-GBS v1 назвал наибольшее количество локусов с большой долей гетерозиготных генотипов (1125), в то время как Stacks назвал только 65 локусов с более чем 50% гетерозигот.
Среди эталонных конвейеров Fast-GBS и TASSEL-GBS v1 назвали наименьшее количество и самые гетерозиготные генотипы (3.4 и 11,5% соответственно). Кроме того, TASSEL-GBS v1 назвал наибольшее количество локусов с большой долей гетерозиготных генотипов (1125), в то время как Stacks назвал только 65 локусов с более чем 50% гетерозигот.
Перекрытие между каталогами SNP
Затем мы определили степень перекрытия между каталогами SNP, полученными с использованием различных конвейеров, и их точность. Мы выбрали Fast-GBS в качестве основы для сравнения из-за его способности очень точно вызывать большое количество SNP.Как показано в , среди эталонных конвейеров наибольшее совпадение наблюдалось между Fast-GBS и Stacks (> 96%), а 92% SNP, вызванных с помощью IGST, также были обнаружены в наборе данных Fast-GBS. Напротив, TASSEL-GBS v1 показал самое низкое перекрытие (36,7%) с Fast-GBS. Конвейеры de novo показали аналогичные уровни перекрытия с Fast-GBS (стеки = 89,1% и UNEAK = 87,5%). В дополнительном анализе (не показанном на рисунке) мы измерили перекрытие между двумя конвейерами de novo ; около 67% SNP, вызванных Stacks, также были обнаружены в наборе данных UNEAK.Таким образом, эти два конвейера de novo , по-видимому, идентифицируют довольно разные подмножества более обширного каталога SNP, полученного с использованием Fast-GBS.
В дополнительном анализе (не показанном на рисунке) мы измерили перекрытие между двумя конвейерами de novo ; около 67% SNP, вызванных Stacks, также были обнаружены в наборе данных UNEAK.Таким образом, эти два конвейера de novo , по-видимому, идентифицируют довольно разные подмножества более обширного каталога SNP, полученного с использованием Fast-GBS.
Таблица 3
Таблица 3
Степень перекрытия среди SNP Loci называется с помощью Fast-GBS и шести других биоинформатических трубопроводов
| Snps | |||||
|---|---|---|---|---|---|
| Подход | Pointure | Общее (в %) | Только другие трубопроводы | Только Fast-GBS | |
| de novo | Стеки | 7 | 13,1 | 1450 | 23100 |
| UNEAK | 24743 | 87,5 | 3172 | 13382 | |
| Ссылка на основе | TASSEL-ГБС v1 | 54412 | 36,7 | 34420 | 14.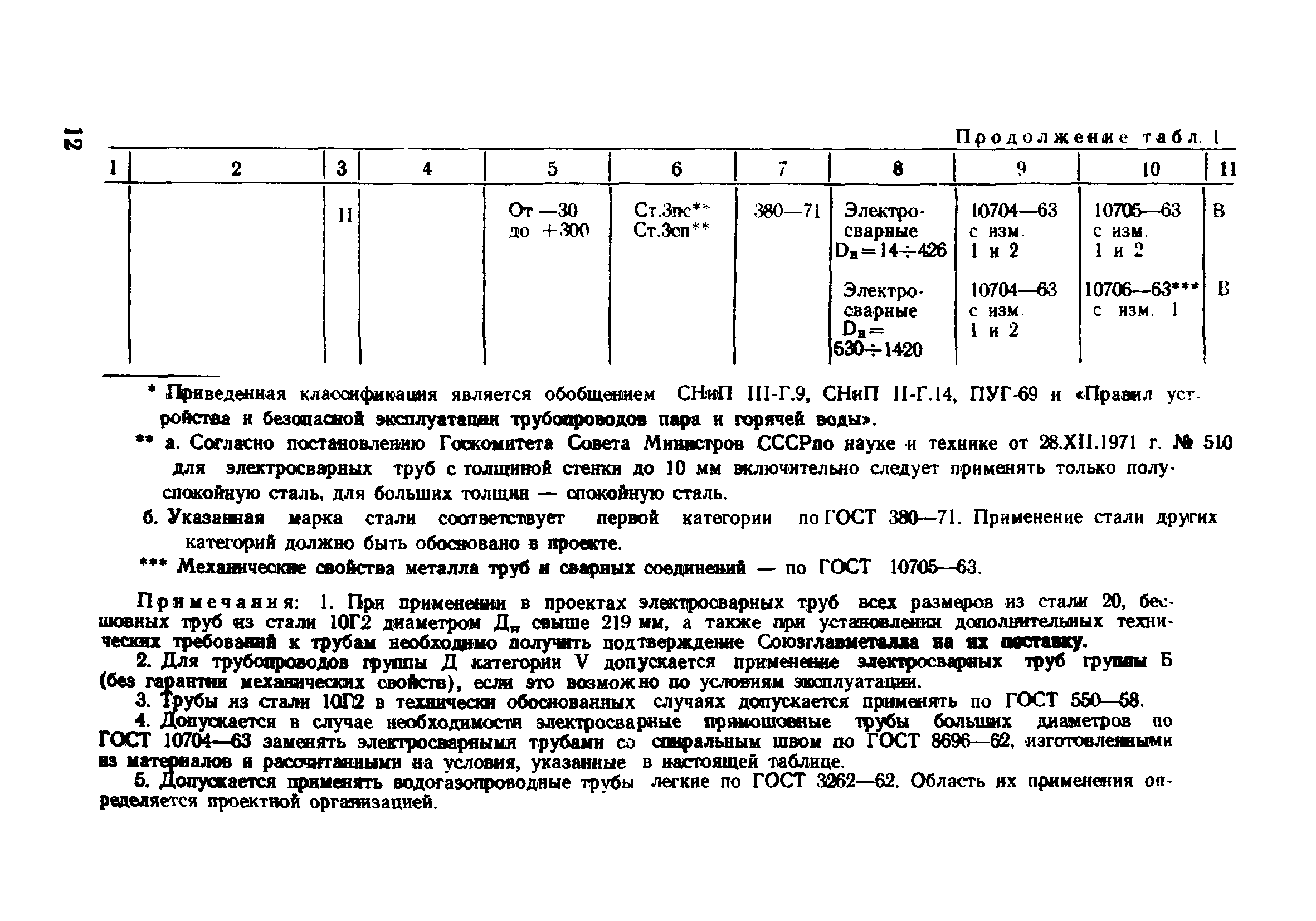 961 961 |
| Стеки | 18941 | 96,2 | 1709 | 16721 | |
| IGST | 25650 | 92,4 | +1950 | 11253 | |
| TASSEL-GBS v2 | 28.158 | 88.3 | 3,295 | 10,090 | |
Чтобы получить более глубокое понимание генотипической точности среди различных подмножеств общих или уникальных SNP, мы подготовили две отдельные диаграммы Венна, каждая из которых включает только четыре конвейера (для ясности), с Fast- GBS включены в обе панели (). Что выделяется на этом рисунке, так это то, что SNP, вызываемые более чем одним конвейером, обычно были очень точными (средневзвешенная точность = 94,8%). Напротив, за единственным исключением Fast-GBS, SNP, вызываемые одним конвейером, обычно были гораздо менее точными (средневзвешенная точность = 66.3%). Наиболее поразительно отметим, что TASSEL-GBS v1 назвал очень большое количество уникальных SNP (более 30 000), которые показывают низкую точность (65%). Уникальные SNP, вызываемые другими пайплайнами, также обычно демонстрировали низкую точность, но их было гораздо меньше, и поэтому в целом они оказывали меньшее влияние.
Что выделяется на этом рисунке, так это то, что SNP, вызываемые более чем одним конвейером, обычно были очень точными (средневзвешенная точность = 94,8%). Напротив, за единственным исключением Fast-GBS, SNP, вызываемые одним конвейером, обычно были гораздо менее точными (средневзвешенная точность = 66.3%). Наиболее поразительно отметим, что TASSEL-GBS v1 назвал очень большое количество уникальных SNP (более 30 000), которые показывают низкую точность (65%). Уникальные SNP, вызываемые другими пайплайнами, также обычно демонстрировали низкую точность, но их было гораздо меньше, и поэтому в целом они оказывали меньшее влияние.
Проценты показывают предполагаемую точность для всех групп SNP (уникальных или общих).
Причины низкой производительности некоторых конвейеров
Учитывая наблюдаемую разницу в количестве вызываемых SNP и их точности, мы решили исследовать причины ошибочных вызовов.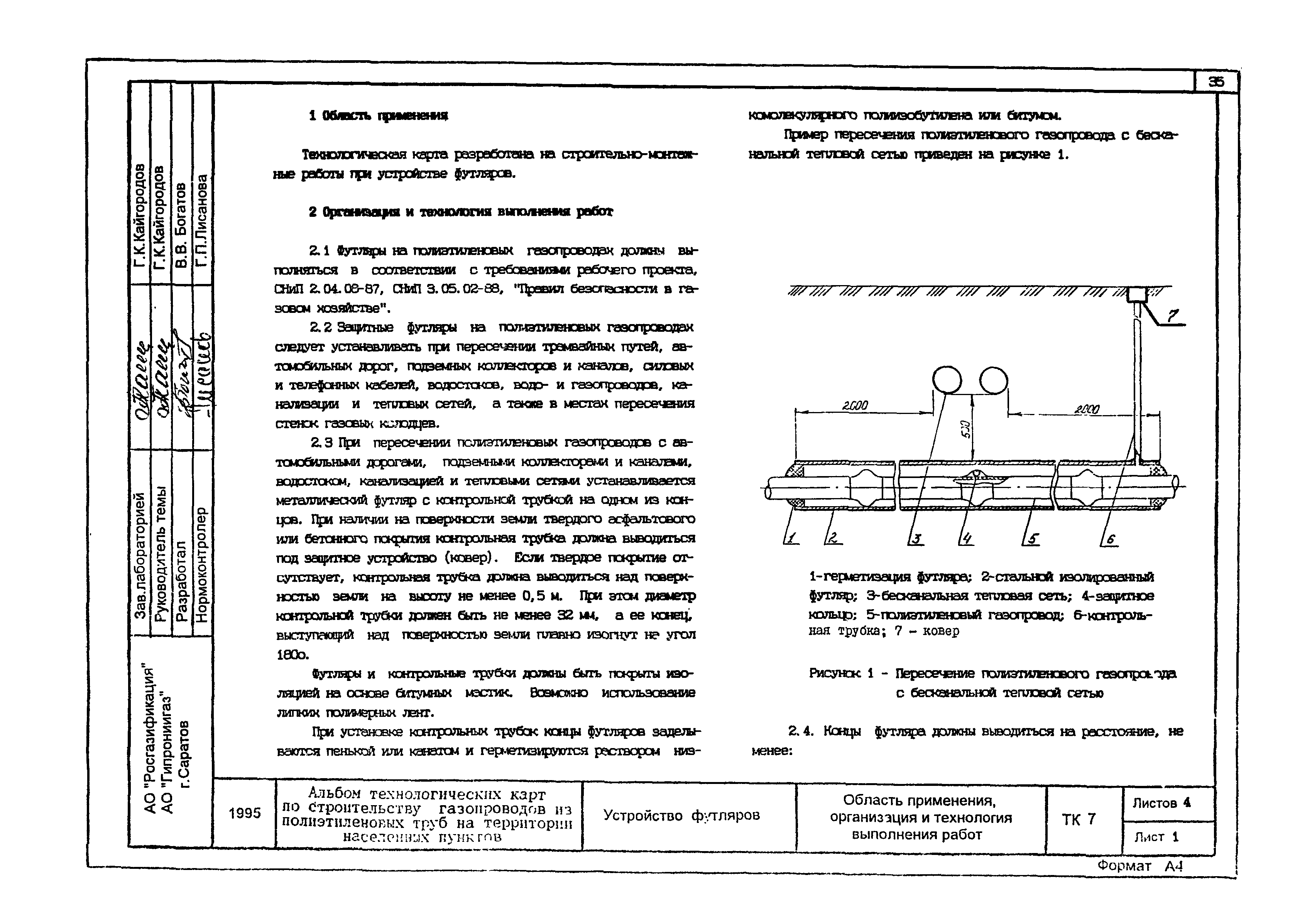 Для проведения этого исследования мы следовали систематическому подходу, показанному на рис. Мы разделили каталог SNP на две категории, точные и неточные, на основе сравнения вызовов, полученных в результате GBS, и вызовов, полученных в результате WGS. Затем неточные SNP были классифицированы либо как уникальные для одного конвейера, либо как общие как минимум для двух конвейеров. Чтобы исследовать уникальные «слабые места» пайплайнов, мы сосредоточили наше внимание на уникальных неточных SNP. Первым шагом в этом исследовании было классифицировать эти неточные SNP как поддерживаемые считываниями, отображаемыми в уникальное положение в геноме, или считываниями, отображаемыми в нескольких позициях.В первом случае ошибки генотипирования были приписаны ошибке вызывающим вариант (например, из-за ошибок секвенирования или амплификации ПЦР). Во втором случае мы пришли к выводу, что картирование прочтений более чем в одном месте генома может быть результатом этих прочтений, происходящих либо из паралогов, либо из повторяющихся областей.
Для проведения этого исследования мы следовали систематическому подходу, показанному на рис. Мы разделили каталог SNP на две категории, точные и неточные, на основе сравнения вызовов, полученных в результате GBS, и вызовов, полученных в результате WGS. Затем неточные SNP были классифицированы либо как уникальные для одного конвейера, либо как общие как минимум для двух конвейеров. Чтобы исследовать уникальные «слабые места» пайплайнов, мы сосредоточили наше внимание на уникальных неточных SNP. Первым шагом в этом исследовании было классифицировать эти неточные SNP как поддерживаемые считываниями, отображаемыми в уникальное положение в геноме, или считываниями, отображаемыми в нескольких позициях.В первом случае ошибки генотипирования были приписаны ошибке вызывающим вариант (например, из-за ошибок секвенирования или амплификации ПЦР). Во втором случае мы пришли к выводу, что картирование прочтений более чем в одном месте генома может быть результатом этих прочтений, происходящих либо из паралогов, либо из повторяющихся областей.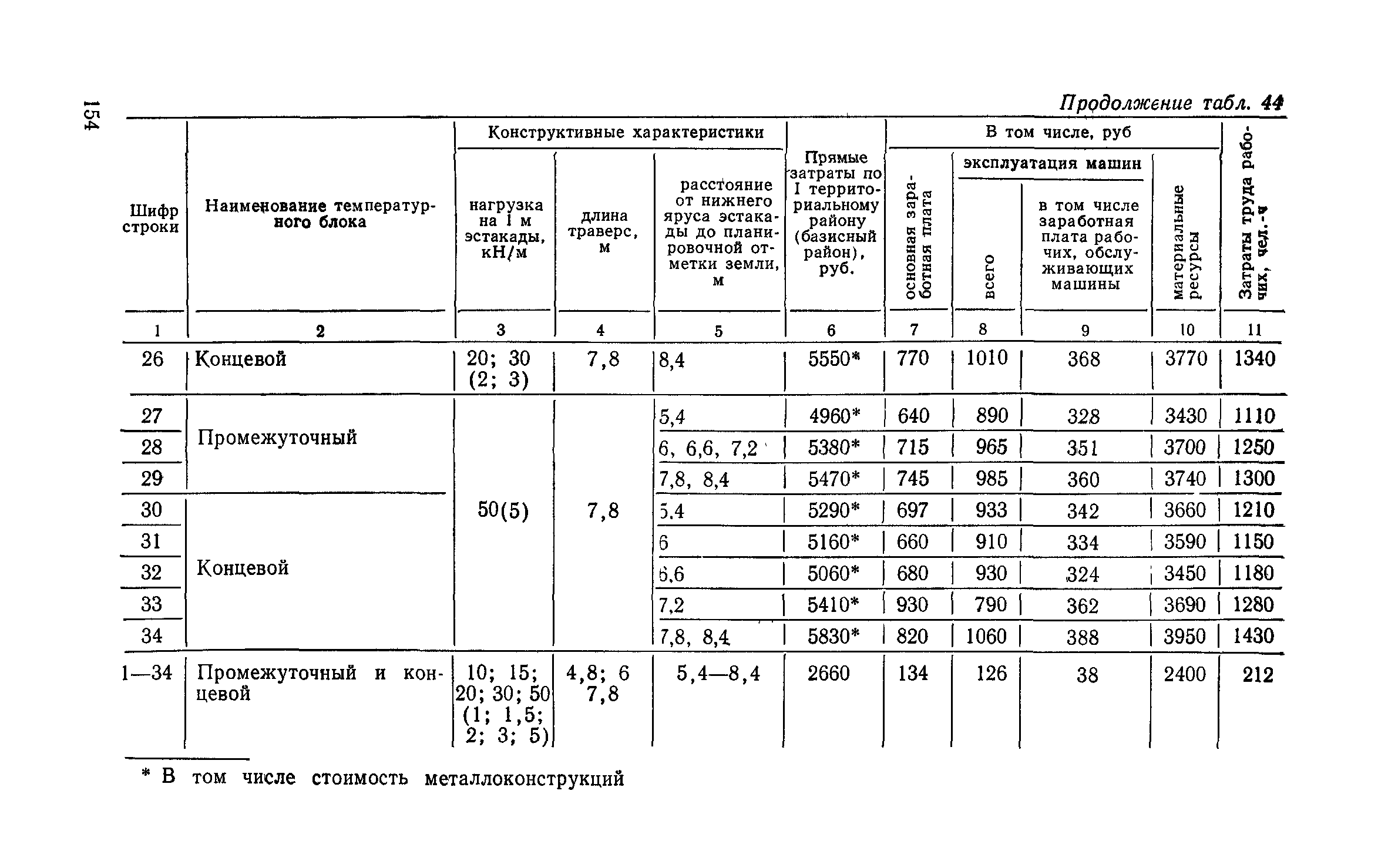 Чтобы решить эту проблему, мы сопоставили чтения с замаскированным эталонным геномом (Phytozome V9: Gmax-189-hardmasked.fa), чтобы оценить долю неточных SNP, происходящих из повторяющихся областей.SNP, которые больше не присутствовали в каталоге, полученном в результате картирования замаскированного эталонного генома, считались связанными с повторяющимися последовательностями. Оставшиеся прочтения, которые успешно сопоставили с несколькими сайтами в замаскированном эталонном геноме, были проанализированы с помощью поиска BLAST для обнаружения паралогии. Чтение считалось производным от паралога, когда мы встречали как минимум 2 совпадения со 100% охватом и минимум 96% идентичности. В среднем считывания, происходящие из паралогичных локусов (как определено выше), имели 2.4 попадания в геном.
Чтобы решить эту проблему, мы сопоставили чтения с замаскированным эталонным геномом (Phytozome V9: Gmax-189-hardmasked.fa), чтобы оценить долю неточных SNP, происходящих из повторяющихся областей.SNP, которые больше не присутствовали в каталоге, полученном в результате картирования замаскированного эталонного генома, считались связанными с повторяющимися последовательностями. Оставшиеся прочтения, которые успешно сопоставили с несколькими сайтами в замаскированном эталонном геноме, были проанализированы с помощью поиска BLAST для обнаружения паралогии. Чтение считалось производным от паралога, когда мы встречали как минимум 2 совпадения со 100% охватом и минимум 96% идентичности. В среднем считывания, происходящие из паралогичных локусов (как определено выше), имели 2.4 попадания в геном.
Систематический подход, используемый для исследования возможных причин уникальных неточных вызовов SNP.
Результаты этого анализа показаны на . Поскольку большинство пайплайнов предоставили в основном точный (>92%) набор SNP, каждый пайплайн вызывал лишь несколько сотен уникальных неточных SNP, за исключением TASSEL-GBS v1 (9828 уникальных неточных SNP).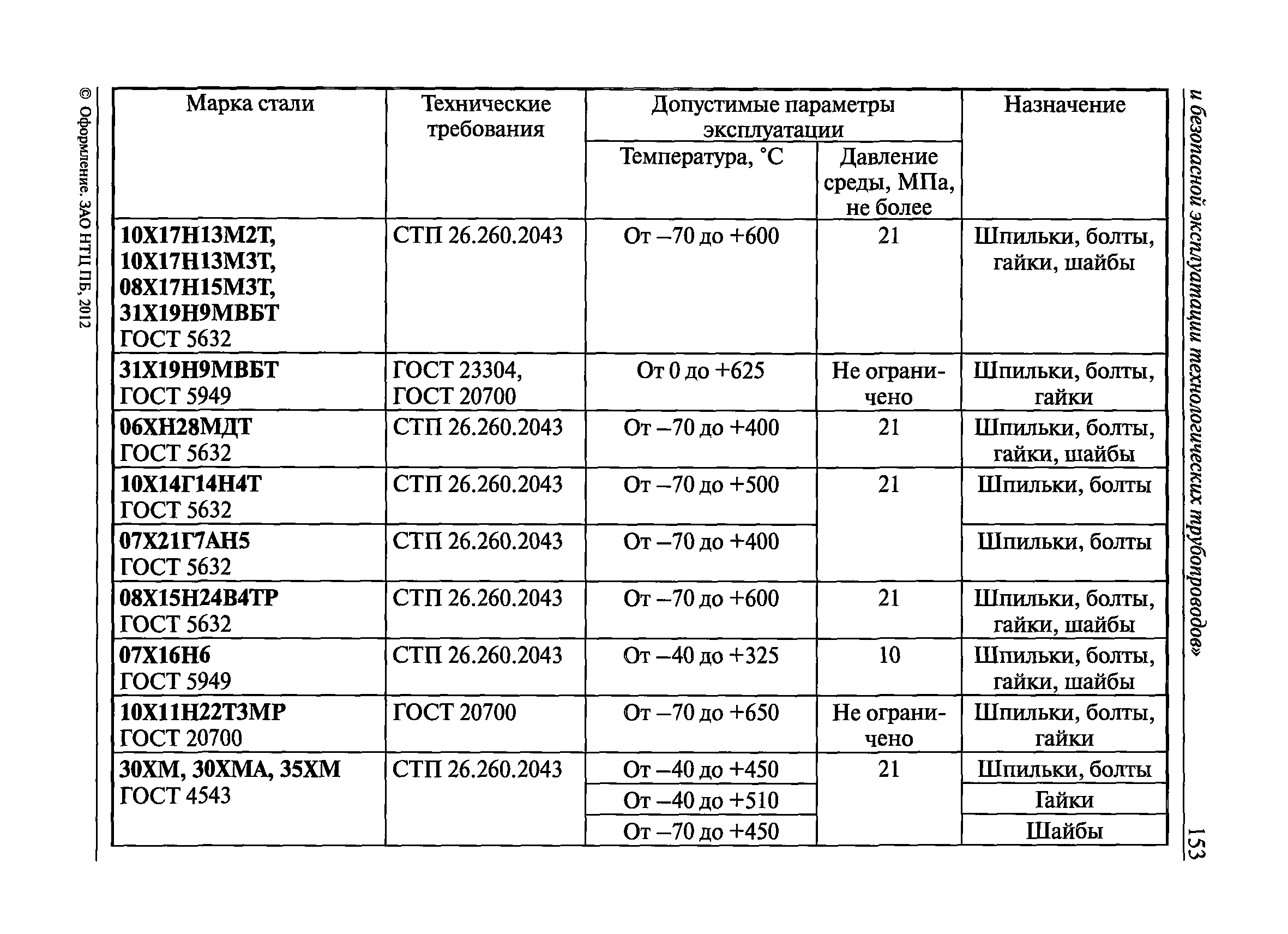 Меньшая часть (от 11,5 до 29,7%) уникальных неточных SNP была подтверждена картированием ридов в одну позицию в геноме и считалась результатом ошибки в вызове вариантов.Большинство (от 70,3 до 88,5%) неточных SNP были подтверждены картированием прочтений более чем в одной области генома. Среди них подавляющее большинство было связано с картированием прочтений в паралогические области (от 74 до 93%). Таким образом, мы пришли к выводу, что большинство ошибок генотипирования сои можно отнести к присутствию паралогов, и что TASSEL-GBS v1 оказался, безусловно, конвейером, наиболее подверженным ошибочным вызовам из-за этого.
Меньшая часть (от 11,5 до 29,7%) уникальных неточных SNP была подтверждена картированием ридов в одну позицию в геноме и считалась результатом ошибки в вызове вариантов.Большинство (от 70,3 до 88,5%) неточных SNP были подтверждены картированием прочтений более чем в одной области генома. Среди них подавляющее большинство было связано с картированием прочтений в паралогические области (от 74 до 93%). Таким образом, мы пришли к выводу, что большинство ошибок генотипирования сои можно отнести к присутствию паралогов, и что TASSEL-GBS v1 оказался, безусловно, конвейером, наиболее подверженным ошибочным вызовам из-за этого.
Таблица 4
Количество и характеристики уникальных неточных SNP, вызванных разными пайплайнами.
| подход | De Novo | Ссылка на основе | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pipeline | Стеки | UNEAK | TASSEL GBS v1 | Стеки | IGST | TASSEL GBS v2 | Fast-GBS | |||
| Уникальный неточный ОНП | 495 | 533 | 9828 | 103 | 207 | 558 | 272 | |||
(3. 7% из 13 303) 7% из 13 303) | (2,2% от 24 743) | (18,1% от 54 412) | (0,5% от 18 941) | (0,8% от 25 650) | (2,0% от 28 158) | (0,8% из 34,953) | ||||
| неточные SNP с уникальной позицией (% от уникальных неточных SNPS) | 146 | 72 | 1 126 | 20 | 46 | 132 | 132 | 35 | ||
(29.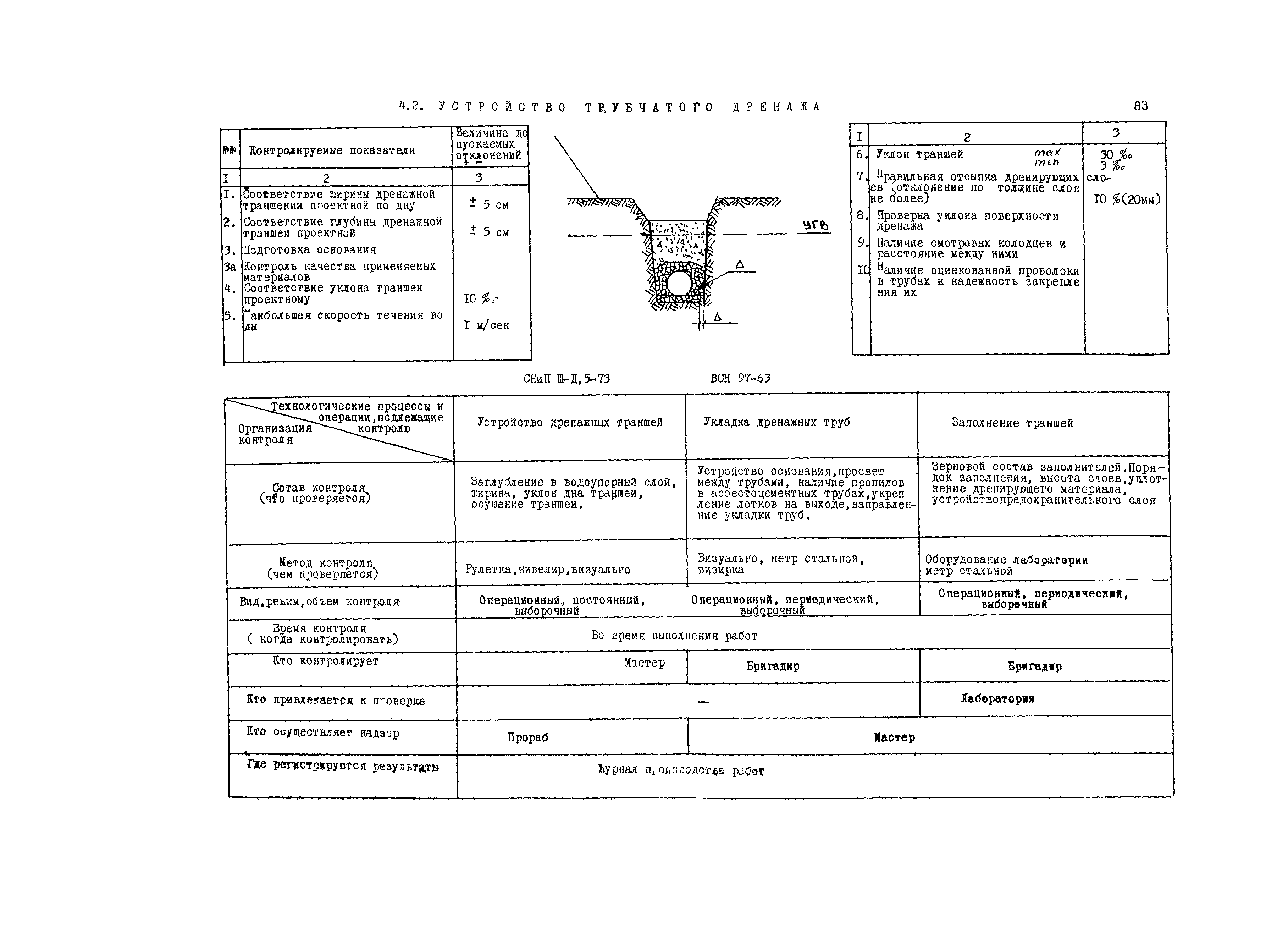 7 ) 7 ) | (13,5) | (11.5) | (19.4) | (22.2) | (23.7) | (12.9) | (12.9) | |||
| неточные Snps с несколькими позициями (% уникальных неточных SNP) | 461 | 461 | 8,702 | 83 | 83 | 161 | 426 | 426 | 237 | |
(70.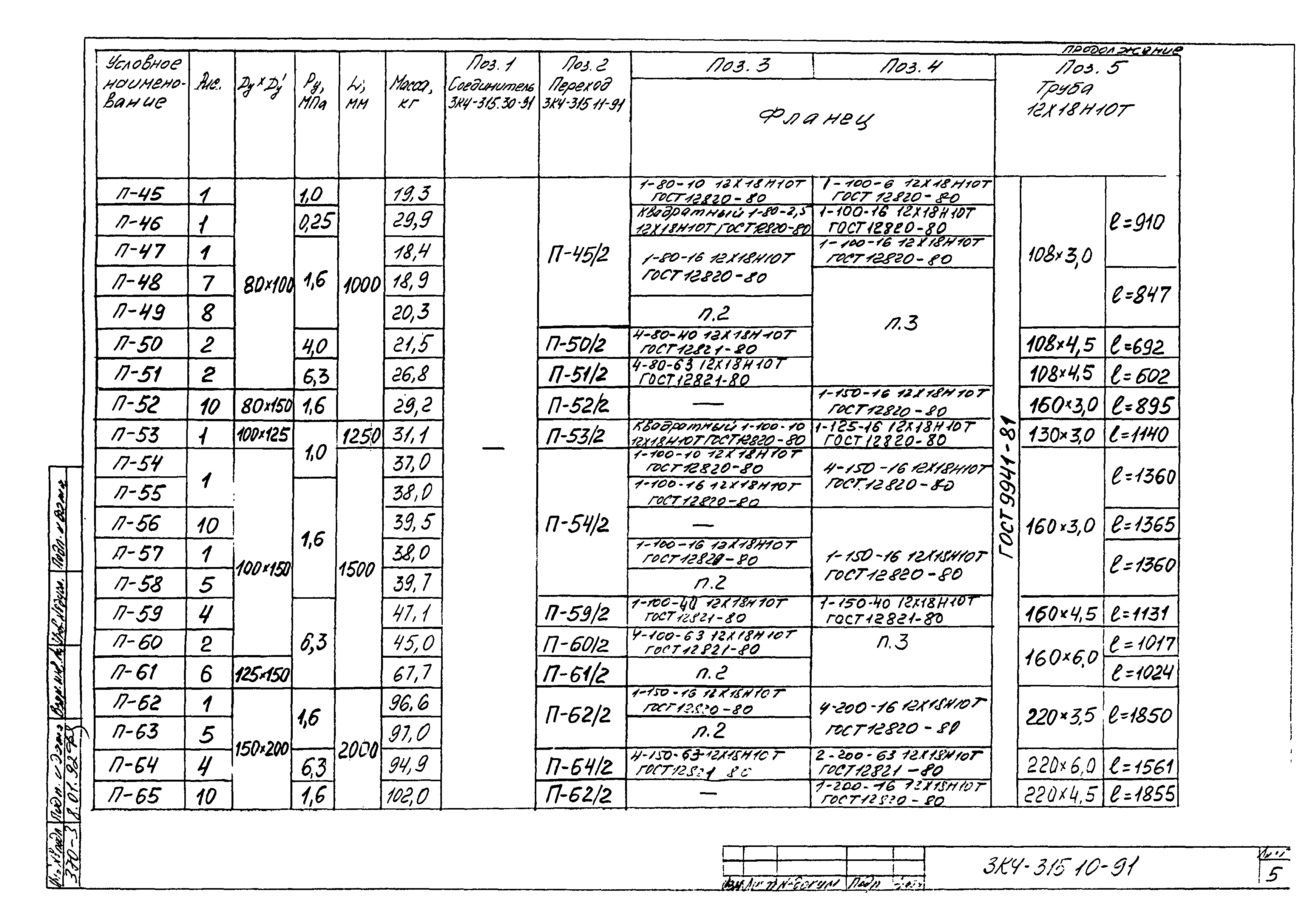 3) 3) | (86.5) | (88.5) | (88.5) | (80.6) | (77.8) | (76.3) | 2 | (87.1) | ||
| повторяющихся региона (% неточных Snps с несколькими позициями) | 45 | 120 | 9 | 7 | 15 | 60872 | 7 17 | |||
| (13) | (13) | (13) | 26) | (21) | (11) | (9) | (14) | (7) | (7) | |
| Paralogues (% неточных Snps с несколькими позициями) | 304 | 34 1 | 6 875 | 7 6 875 | 74 | 146 | 146 | 366 | 220 | 220 |
| (74) | (74) | (79) | (89) | (91) | (86) | (86) | (93) | |||
Другим результатом, который требовал исследования, было относительно небольшое количество SNP, вызванных Stacks, поскольку и de novo , и основанные на эталонах версии Stacks вызывали наименьшее количество SNP. Мы исследовали эффективность шага демультиплексирования, поскольку он уже был описан как проблематичный. В нашем анализе мы обнаружили, что 19,7% считываний Illumina не удалось присвоить конкретному файлу штрих-кода, что намного выше, чем у других конвейеров. Чтобы измерить влияние такого уменьшения количества чтений, доступных для вызова SNP, мы использовали альтернативный инструмент демультиплексирования (Sabre) вместо того, который предоставляется в Stacks. Доля пропущенных прочтений уменьшилась до ~ 2%, а количество SNP, вызванных с использованием этого более обширного набора прочтений, увеличилось на 12 и 24% (21 456 и 17 342) для стеков на основе ссылок и стеков de novo соответственно.Мы пришли к выводу, что низкая производительность инструмента демультиплексирования Stacks является важной причиной уменьшения количества SNP, вызываемых Stacks.
Мы исследовали эффективность шага демультиплексирования, поскольку он уже был описан как проблематичный. В нашем анализе мы обнаружили, что 19,7% считываний Illumina не удалось присвоить конкретному файлу штрих-кода, что намного выше, чем у других конвейеров. Чтобы измерить влияние такого уменьшения количества чтений, доступных для вызова SNP, мы использовали альтернативный инструмент демультиплексирования (Sabre) вместо того, который предоставляется в Stacks. Доля пропущенных прочтений уменьшилась до ~ 2%, а количество SNP, вызванных с использованием этого более обширного набора прочтений, увеличилось на 12 и 24% (21 456 и 17 342) для стеков на основе ссылок и стеков de novo соответственно.Мы пришли к выводу, что низкая производительность инструмента демультиплексирования Stacks является важной причиной уменьшения количества SNP, вызываемых Stacks.
GBS с использованием разных платформ секвенирования
Чтобы сравнить вызов SNP с использованием различных технологий секвенирования, мы выполнили GBS на тех же 24 образцах сои на платформе Ion Torrent. В отличие от ридов Illumina, которые имеют одинаковую длину (100 п.н.), риды Ion Torrent имеют длину от 50 до 135 п.н. В этом анализе мы использовали только два эталонных конвейера, которые показали лучшие результаты в описанных выше тестах (Fast-GBS и TASSEL-GBS v2) с использованием 38 миллионов чтений Ion Torrent.Как видно на рисунке, количество SNP, вызванных с каждым конвейером при одинаковых уровнях допуска отсутствующих данных (≤80%) и частоте минорных аллелей (≥0,05), было очень схожим (~23 тыс. в обоих случаях). Как и выше, Fast-GBS назвал большее количество вариантов, так как он назвал в общей сложности более 2000 вставок в дополнение к SNP. По времени вычислений Fast-GBS был более чем в два раза быстрее, чем TASSEL-GBS v2 (1 ч 41 мин против 3 ч 39 мин), при этом он использовал на 15 % больше дискового пространства (20 Гб против 17 Гб).
В отличие от ридов Illumina, которые имеют одинаковую длину (100 п.н.), риды Ion Torrent имеют длину от 50 до 135 п.н. В этом анализе мы использовали только два эталонных конвейера, которые показали лучшие результаты в описанных выше тестах (Fast-GBS и TASSEL-GBS v2) с использованием 38 миллионов чтений Ion Torrent.Как видно на рисунке, количество SNP, вызванных с каждым конвейером при одинаковых уровнях допуска отсутствующих данных (≤80%) и частоте минорных аллелей (≥0,05), было очень схожим (~23 тыс. в обоих случаях). Как и выше, Fast-GBS назвал большее количество вариантов, так как он назвал в общей сложности более 2000 вставок в дополнение к SNP. По времени вычислений Fast-GBS был более чем в два раза быстрее, чем TASSEL-GBS v2 (1 ч 41 мин против 3 ч 39 мин), при этом он использовал на 15 % больше дискового пространства (20 Гб против 17 Гб).
Таблица 5
Таблица 5
Количество Snps и Indels Обнаружены среди 24 соевых линий с использованием ионов Torrent READS и два разных биоинформатических трубопровода
*| варианты | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Трубопровод | SNP | INDEL | Время | (H: M) | память (GB) | ||||||
| Tassel-GBSV2 | 22,921 | ND | 3:29 | 3:29 | 3:29 | 17 | 17 | ||||
| Fast-GBS | 23,792 | 2,054 | 2,054 | 1:31 | 20 | 7 20 | |||||
Во втором анализе мы измерили объем пропущенных данных и оценивающую точность генотипов как путем сравнения GBS -названные генотипы к полученным в результате ресеквенирования и путем оценки степени гетерозиготности в этих линиях, предполагаемых мед гомозигот.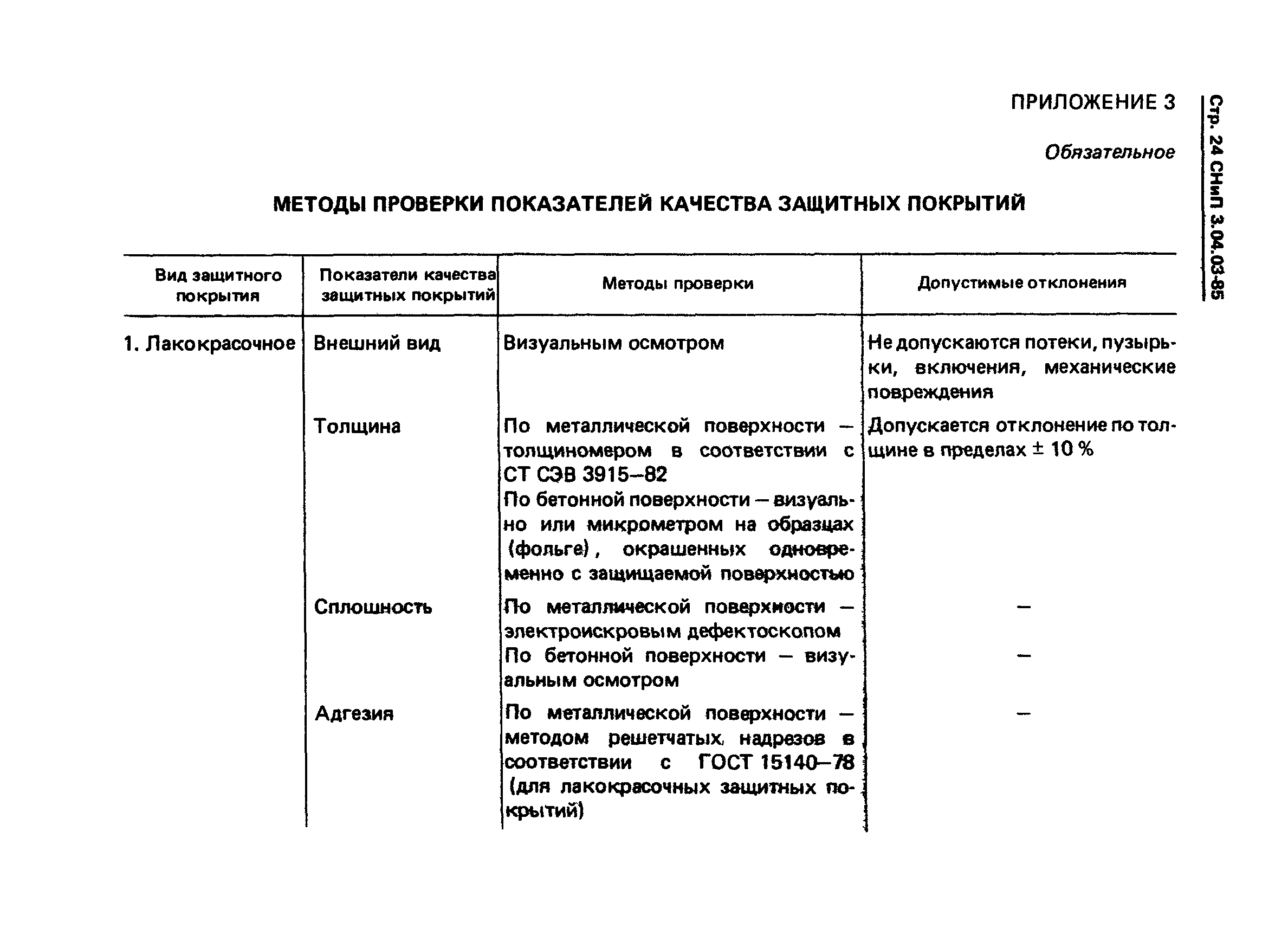 Как видно из рисунка, доля отсутствующих данных была относительно одинаковой для двух конвейеров (37% против 33%). В этом анализе TASSEL-GBS v2 вызвал больше гетерозиготных генотипов, чем Fast-GBS (6,6% против 4,5%). Также TASSEL-GBS v2 назвал гораздо больше локусов с большей долей (> 50%) гетерозиготных генотипов, чем Fast-GBS (4831 против 861). В этом анализе Fast-GBS снова добился наивысшей точности определения генотипов (95,2%) по сравнению с 91,1% при использовании TASSEL-GBS v2.
Как видно из рисунка, доля отсутствующих данных была относительно одинаковой для двух конвейеров (37% против 33%). В этом анализе TASSEL-GBS v2 вызвал больше гетерозиготных генотипов, чем Fast-GBS (6,6% против 4,5%). Также TASSEL-GBS v2 назвал гораздо больше локусов с большей долей (> 50%) гетерозиготных генотипов, чем Fast-GBS (4831 против 861). В этом анализе Fast-GBS снова добился наивысшей точности определения генотипов (95,2%) по сравнению с 91,1% при использовании TASSEL-GBS v2.
Таблица 6
Таблица 6
Точность данных SNP, полученные с использованием ION Torrent READS и двух разных биоинформатических трубопроводов
7 95.2 95.2 95.2| тип статистика / трубопровод | TASSEL-GBSV2 | FAST-GBS | ||||
|---|---|---|---|---|---|---|
| ОНП | 22921 | 23792 | ||||
| Отсутствующие данные (%) | 37 | 33 | ||||
| локусов с> 50% гетерозигот * | 4831 | 861 | ||||
| Остаточные гетерозиготы (%) | 6. 6 6 | 4.5 | 45 | 45 | ||
| Точность (%) | 91.1 | 91.1 | 95.2 | 95.2 | 95.2 | 95.2 |
Как показано на рисунке , при использовании Fast-GBS мы обнаружили, что 69% (16 416 из 23 792 SNP) SNP, полученных из ридов Ion Torrent, также присутствовали в каталоге SNP, полученных с помощью ридов Illumina. И наоборот, из всех SNP, вызванных с помощью чтения Illumina (34 953 SNP), 47% были общими с каталогом Ion Torrent.Используя TASSEL-GBS v2, немного меньшая доля (54%) (12 377 из 22 921 SNP) SNP, вызванных из ридов Ion Torrent, также была получена с использованием ридов Illumina.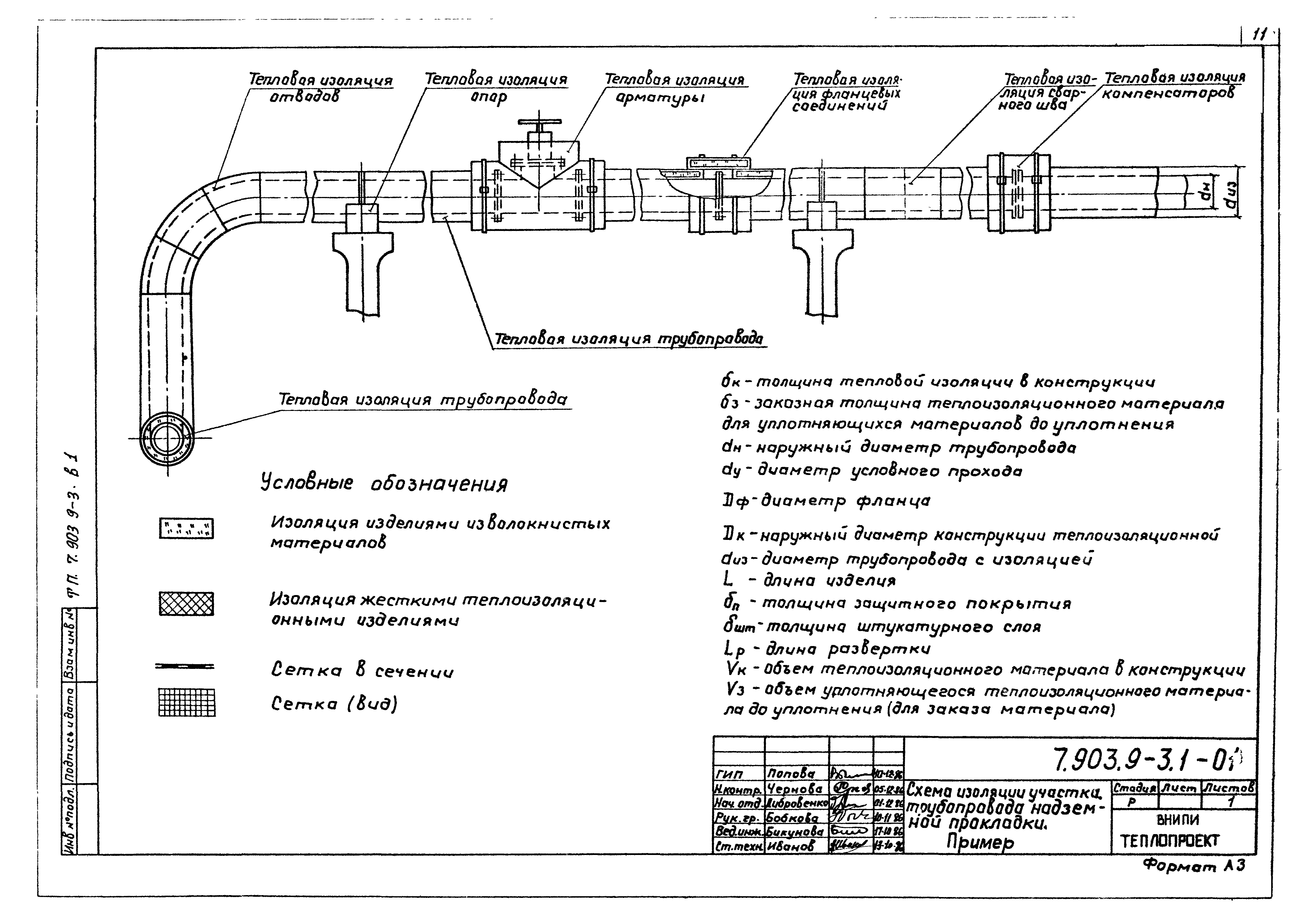 И наоборот, аналогичная доля (44%) SNP, вызванных с использованием ридов Illumina, была общей с теми, которые были вызваны с помощью ридов Ion Torrent. Мы обнаружили, что использование чтения Ion Torrent приводит к большему количеству неточных SNP по сравнению с чтением Illumina. При использовании чтения Illumina только 23,7% и 12,9% неточных SNP, вызванных TASSEL-GBS v2 и Fast-GBS, имели уникальную позицию, в то время как при использовании чтения Ion Torrent эта доля увеличилась до 76% и 87% для TASSEL-GBS v2 и Fast-GBS. ГБС соответственно.С другой стороны, количество неточных SNP из-за паралогии и повторяющихся областей было одинаковым для обеих технологий. Основываясь на этих результатах, мы делаем вывод, что наблюдаемое увеличение количества неточных SNP с уникальным положением (не из-за какой-либо повторяющейся последовательности) связано с более высокой частотой ошибок секвенирования в чтениях Ion Torrent.
И наоборот, аналогичная доля (44%) SNP, вызванных с использованием ридов Illumina, была общей с теми, которые были вызваны с помощью ридов Ion Torrent. Мы обнаружили, что использование чтения Ion Torrent приводит к большему количеству неточных SNP по сравнению с чтением Illumina. При использовании чтения Illumina только 23,7% и 12,9% неточных SNP, вызванных TASSEL-GBS v2 и Fast-GBS, имели уникальную позицию, в то время как при использовании чтения Ion Torrent эта доля увеличилась до 76% и 87% для TASSEL-GBS v2 и Fast-GBS. ГБС соответственно.С другой стороны, количество неточных SNP из-за паралогии и повторяющихся областей было одинаковым для обеих технологий. Основываясь на этих результатах, мы делаем вывод, что наблюдаемое увеличение количества неточных SNP с уникальным положением (не из-за какой-либо повторяющейся последовательности) связано с более высокой частотой ошибок секвенирования в чтениях Ion Torrent.
Диаграмма Венна для перекрытия SNP, вызванных с использованием двух разных конвейеров биоинформатики (a) Перекрытие SNP, вызванных с помощью Fast-GBS с использованием чтения Illumina и Ion Torrent.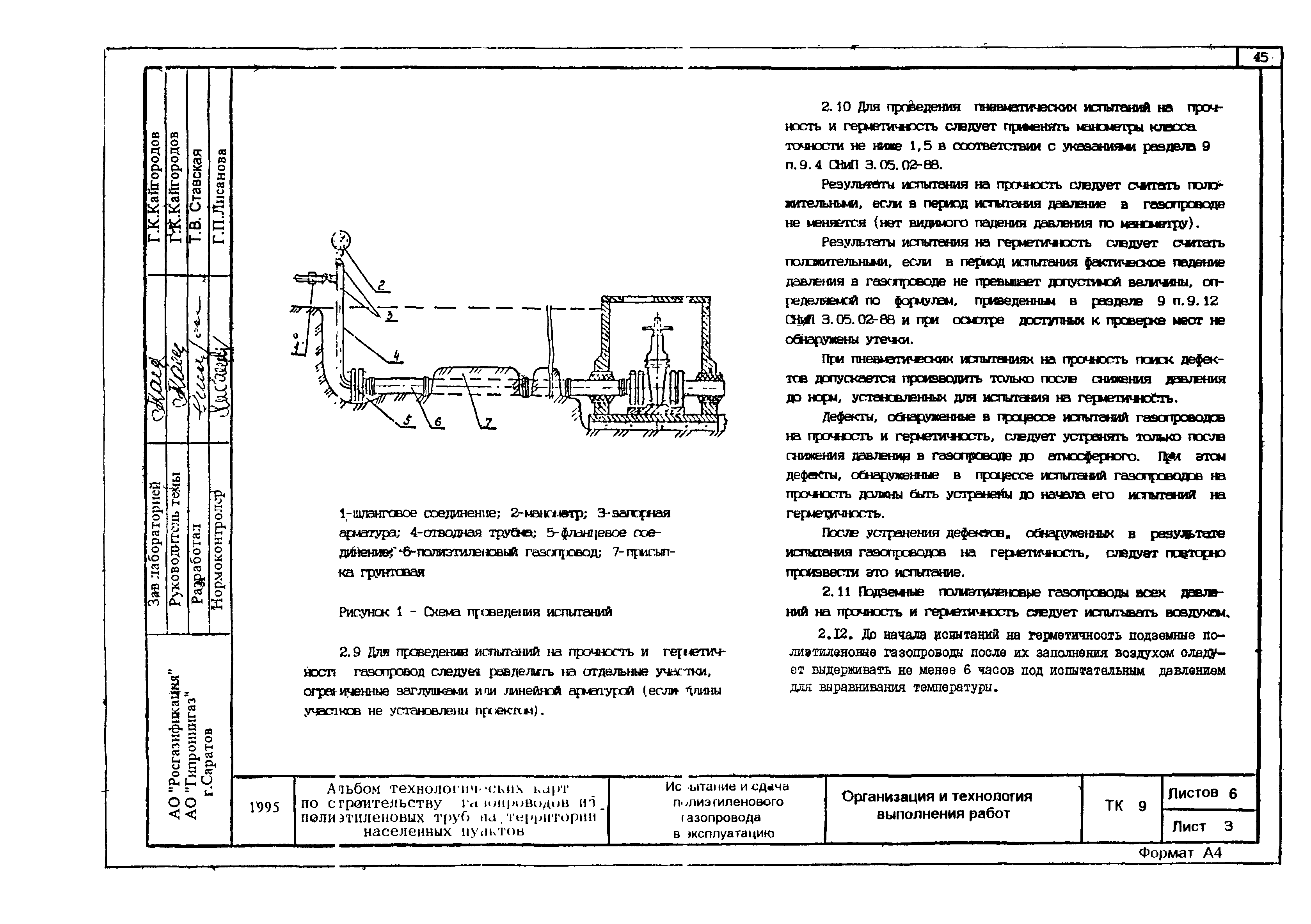 (b) Перекрытие SNP, вызванных с помощью TASSEL-GBS v2 с использованием чтения Illumina и Ion Torrent. Проценты указывают предполагаемую точность для всех групп SNP (уникальных или общих).
(b) Перекрытие SNP, вызванных с помощью TASSEL-GBS v2 с использованием чтения Illumina и Ion Torrent. Проценты указывают предполагаемую точность для всех групп SNP (уникальных или общих).
В заключение, степень перекрытия между платформами секвенирования была одинаковой при использовании обоих конвейеров, но намного ниже, чем перекрытие между конвейерами, использующими одну и ту же платформу секвенирования.
Обсуждение
Гибкость и низкая стоимость методов генотипирования, основанных на NGS, делают эти превосходные инструменты для многих приложений и исследовательских вопросов в области генетики, селекции и биоразнообразия [3, 6, 23–25].В настоящее время GBS, по-видимому, предпочтительнее в сельскохозяйственных науках (селекция растений и животных), тогда как RAD-Seq, по-видимому, является более распространенным подходом в области экологии [1]. Какой бы подход к подготовке библиотеки ни был выбран для снижения сложности перед секвенированием, необходимо использовать биоинформатику для извлечения полезной информации о локусах SNP и генотипах из огромного количества ридов коротких последовательностей [1, 26]. Именно на этом этапе выбор аналитического метода будет иметь наибольшее влияние на количество и качество получаемой генотипической информации.К сожалению, на сегодняшний день в нескольких исследованиях систематически сравнивались конвейеры вызова SNP для GBS и сравнивалась их эффективность, точность и степень перекрытия.
Именно на этом этапе выбор аналитического метода будет иметь наибольшее влияние на количество и качество получаемой генотипической информации.К сожалению, на сегодняшний день в нескольких исследованиях систематически сравнивались конвейеры вызова SNP для GBS и сравнивалась их эффективность, точность и степень перекрытия.
Первый вопрос, который возникает, касается использования de novo по сравнению с эталонными методами. В отсутствие эталонного генома нет другого выбора, кроме как использовать один из двух широко распространенных в настоящее время инструментов: UNEAK и Stacks. Хотя для этого используются разные алгоритмы, эти два конвейера концептуально схожи в том, что они стремятся сначала установить каталоги идентичных прочтений, а затем искать тесно связанные прочтения, которые потенциально являются аллелями в одном и том же локусе.В условиях, используемых в этой работе, UNEAK значительно превзошел Stacks в том, что он генерировал на 82% больше SNP (~ 25 тыс. против ~ 13 тыс. ). С качественной точки зрения оба пайплайна de novo показали себя одинаково хорошо с точки зрения отсутствующих данных (~40%) и генотипической точности (~94%). Это сопоставимо с результатами, полученными Lu et al. (2013) на кукурузе, где было подсчитано, что 92% вызовов генотипа были точными и что эта доля может быть увеличена до 96,2% путем фильтрации SNP с MAF > 0.3 в сегрегирующей биродительской популяции [16]. Оба конвейера de novo могут работать довольно быстро и относительно консервативны в своих вызовах SNP, что приводит к набору данных высокого качества. Таким образом, для подавляющего большинства видов, для которых нет эталонного генома в настоящее время или в обозримом будущем, инструменты вызова SNP de novo работают очень хорошо с точки зрения точности, но UNEAK даст почти в два раза больше SNP.
). С качественной точки зрения оба пайплайна de novo показали себя одинаково хорошо с точки зрения отсутствующих данных (~40%) и генотипической точности (~94%). Это сопоставимо с результатами, полученными Lu et al. (2013) на кукурузе, где было подсчитано, что 92% вызовов генотипа были точными и что эта доля может быть увеличена до 96,2% путем фильтрации SNP с MAF > 0.3 в сегрегирующей биродительской популяции [16]. Оба конвейера de novo могут работать довольно быстро и относительно консервативны в своих вызовах SNP, что приводит к набору данных высокого качества. Таким образом, для подавляющего большинства видов, для которых нет эталонного генома в настоящее время или в обозримом будущем, инструменты вызова SNP de novo работают очень хорошо с точки зрения точности, но UNEAK даст почти в два раза больше SNP.
Картина производительности трубопроводов de novo в этом сравнении может быть слишком радужной.Действительно, ради единообразия мы использовали одни и те же параметры фильтрации (MinMAF≥0,05, MaxMD = 80% и minDP≥2) как для de novo , так и для эталонных конвейеров.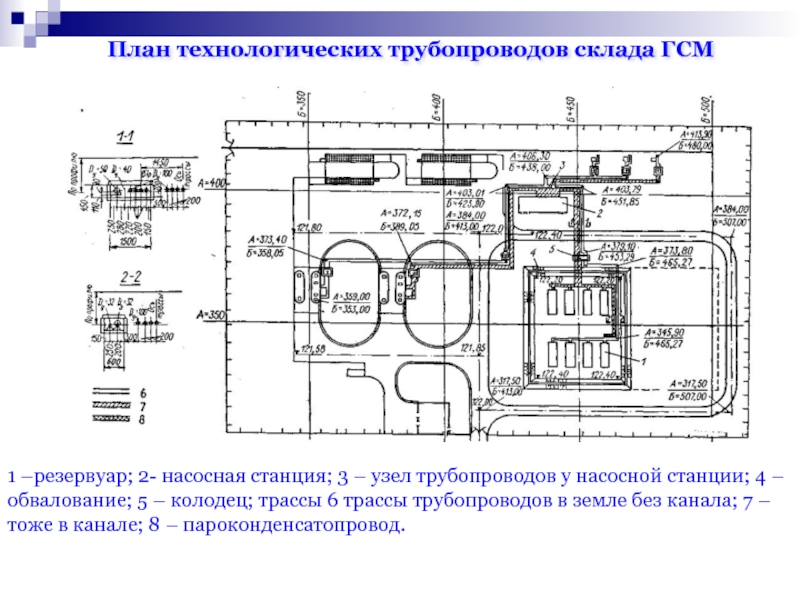 Но такая высокая устойчивость к отсутствующим данным может оказаться нереалистичной в случае конвейеров de novo . Ранее мы показали, что импутация отсутствующих данных очень эффективна и точна для плотного набора SNP, полученного с использованием эталонного конвейера [18]. В случае конвейеров de novo при отсутствии информации о местоположении различных SNP и структуре гаплотипов вменение является гораздо более сложной задачей.По этой причине большинство пользователей конвейеров de novo устанавливают более низкий потолок для максимального объема недостающих данных, обычно между 20% и не более 50% [16, 19, 27]. При использовании данных последовательности GBS, используемых в этой работе, допуск до 20% отсутствующих данных существенно снижает количество SNP, которые можно вызвать с использованием обоих конвейеров de novo (~ 5 тыс. SNP; данные не показаны). В этих более реалистичных условиях (с учетом необходимого вменения отсутствующих данных) мы обнаружили, что эталонные пайплайны дали примерно в 5–7 раз больше высококачественных маркеров SNP (~ 5 тыс.
Но такая высокая устойчивость к отсутствующим данным может оказаться нереалистичной в случае конвейеров de novo . Ранее мы показали, что импутация отсутствующих данных очень эффективна и точна для плотного набора SNP, полученного с использованием эталонного конвейера [18]. В случае конвейеров de novo при отсутствии информации о местоположении различных SNP и структуре гаплотипов вменение является гораздо более сложной задачей.По этой причине большинство пользователей конвейеров de novo устанавливают более низкий потолок для максимального объема недостающих данных, обычно между 20% и не более 50% [16, 19, 27]. При использовании данных последовательности GBS, используемых в этой работе, допуск до 20% отсутствующих данных существенно снижает количество SNP, которые можно вызвать с использованием обоих конвейеров de novo (~ 5 тыс. SNP; данные не показаны). В этих более реалистичных условиях (с учетом необходимого вменения отсутствующих данных) мы обнаружили, что эталонные пайплайны дали примерно в 5–7 раз больше высококачественных маркеров SNP (~ 5 тыс.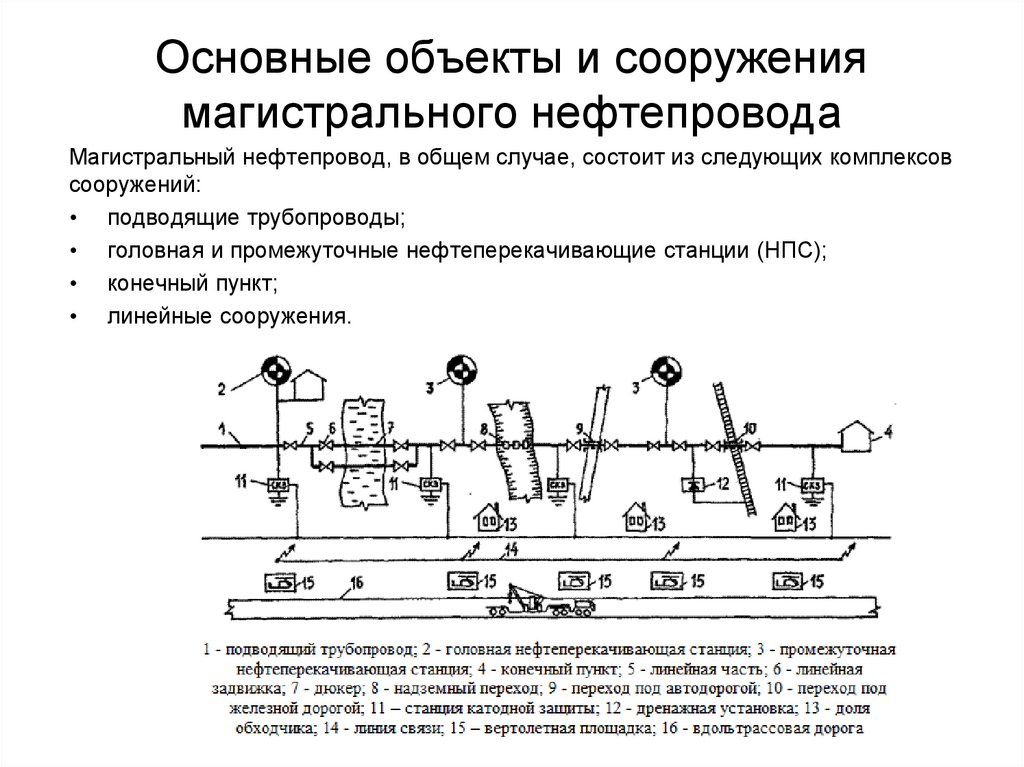 маркеров против 25 тыс.–35 тыс.).
маркеров против 25 тыс.–35 тыс.).
Учитывая растущую доступность эталонных геномов экономически важных сельскохозяйственных культур и животных, нам необходимо задаться вопросом, какой из доступных эталонных конвейеров дает лучший каталог SNP как с точки зрения изобилия маркеров, так и с точки зрения их точности. Среди пяти конвейеров, основанных на эталонах, Fast-GBS можно запустить быстро, что привело к самой высокой точности генотипирования для очень большого количества локусов SNP (около 35 000) в дополнение к почти 4000 инделей. Исходя из этих соображений, он кажется предпочтительным, по крайней мере, в случае сои и, вероятно, также для других видов с аналогичными геномными и репродуктивными характеристиками.
Из протестированных пайплайнов TASSEL-GBSv1 выделялся из остальной группы по количеству вызываемых локусов SNP (на 50–100 % больше, чем у других), но это происходило за счет точности, поскольку это был единственный трубопровод, чьи генотипические вызовы были точными менее чем в 90% случаев (76,1%). Поскольку отличить истинный генотип от ложного непросто, мы утверждаем, что TASSEL-GBSv1 недостаточно точен, чтобы его можно было использовать отдельно. В предыдущей работе большой результирующий каталог SNP часто «фильтровался» путем отбрасывания маркеров, которые не вели себя должным образом в сегрегирующей популяции [6].Это, по-видимому, помогло отбросить «ложные» маркеры, возникшие в результате смешения аллелей (в одном локусе) и прочтений, полученных из паралогичных локусов. Мы предположили, что основной причиной снижения точности является тот факт, что TASSEL-GBSv1 обрезает все чтения до одинаковой длины в 64 базы, создавая таким образом короткие теги, которые подвергаются повышенному риску сопоставления с несколькими или ошибочными местоположениями. Конвейеры, использующие более длительные чтения, не проявляли этой проблемы и обычно имели как минимум в 10 раз меньше операций чтения, сопоставленных с несколькими местоположениями.Например, несмотря на то, что у TASSEL-GBS v1 много общего, когда TASSEL-GBS v2 запускали в условиях, позволяющих использовать более длинные теги (в нашем случае 92 основания), надежность генотипов значительно возросла.
Поскольку отличить истинный генотип от ложного непросто, мы утверждаем, что TASSEL-GBSv1 недостаточно точен, чтобы его можно было использовать отдельно. В предыдущей работе большой результирующий каталог SNP часто «фильтровался» путем отбрасывания маркеров, которые не вели себя должным образом в сегрегирующей популяции [6].Это, по-видимому, помогло отбросить «ложные» маркеры, возникшие в результате смешения аллелей (в одном локусе) и прочтений, полученных из паралогичных локусов. Мы предположили, что основной причиной снижения точности является тот факт, что TASSEL-GBSv1 обрезает все чтения до одинаковой длины в 64 базы, создавая таким образом короткие теги, которые подвергаются повышенному риску сопоставления с несколькими или ошибочными местоположениями. Конвейеры, использующие более длительные чтения, не проявляли этой проблемы и обычно имели как минимум в 10 раз меньше операций чтения, сопоставленных с несколькими местоположениями.Например, несмотря на то, что у TASSEL-GBS v1 много общего, когда TASSEL-GBS v2 запускали в условиях, позволяющих использовать более длинные теги (в нашем случае 92 основания), надежность генотипов значительно возросла.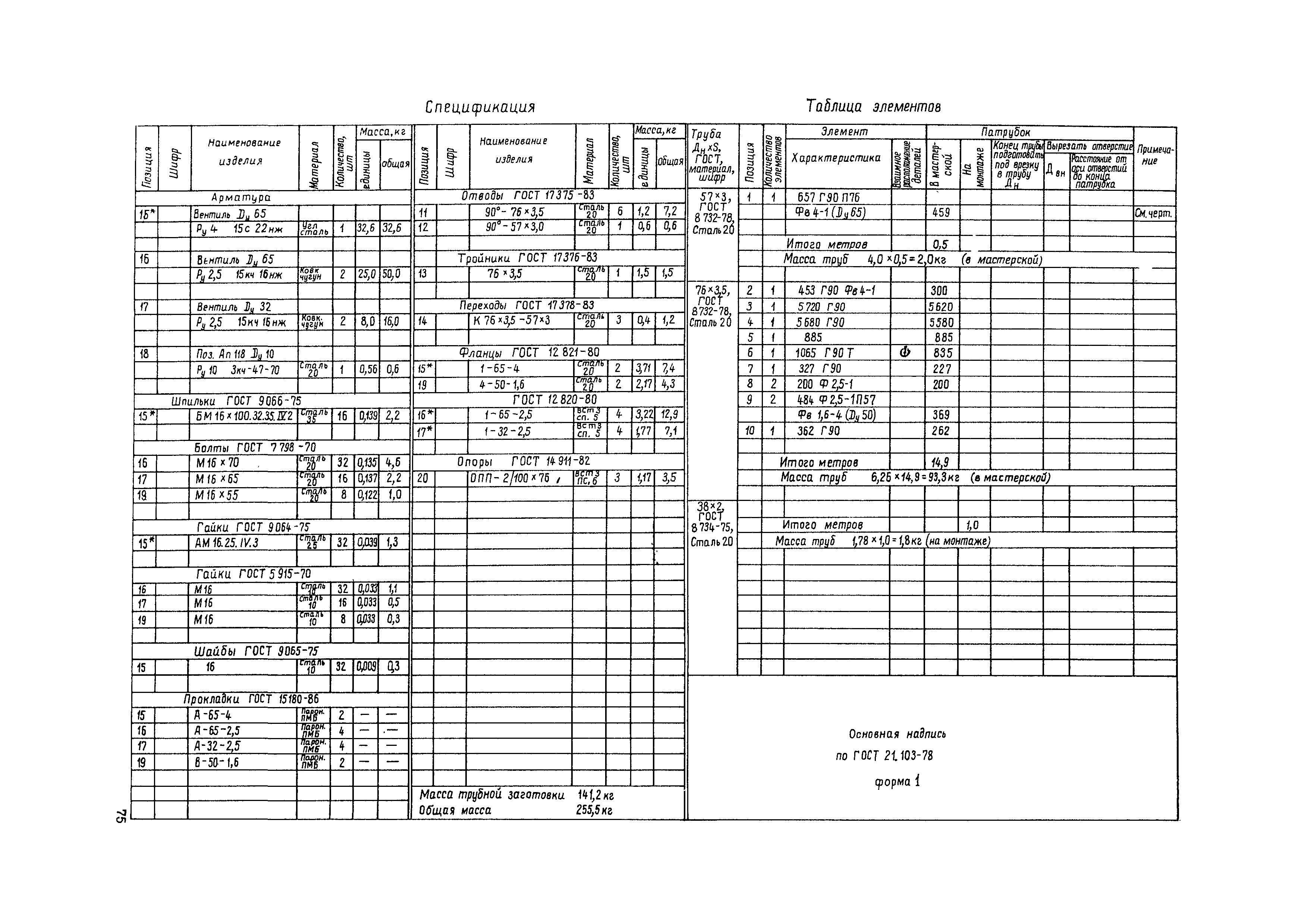
Основанная на ссылках версия Stacks — это другой конвейер, который выделяется тем, что вызывает гораздо меньше SNP, чем другие. При исследовании различных шагов, необходимых для перехода от последовательностей к SNP, мы обнаружили, что стеки потеряли ~ 20% чтений на этапе демультиплексирования, т.е.некоторые считывания штрих-кода не относились к образцу и просто отбрасывались на последующих этапах. Это, очевидно, привело к сопутствующему уменьшению количества названных SNP (~ 19 тыс. против ~ 25 тыс.). Об этой плохой производительности шага демультиплексирования Stacks ранее сообщалось Хертеном и др. [28].
По нашему мнению, полногеномное измерение точности наборов данных GBS, полученных из различных каналов биоинформатики, представляет собой важный и ключевой вклад в эту работу. Его оценивали путем прямого сравнения с данными ресеквенирования всего генома.Во многих предыдущих исследованиях оценка генотипической точности часто достигалась косвенным измерением [16] или выполнялась на очень небольшом подмножестве локусов SNP [9].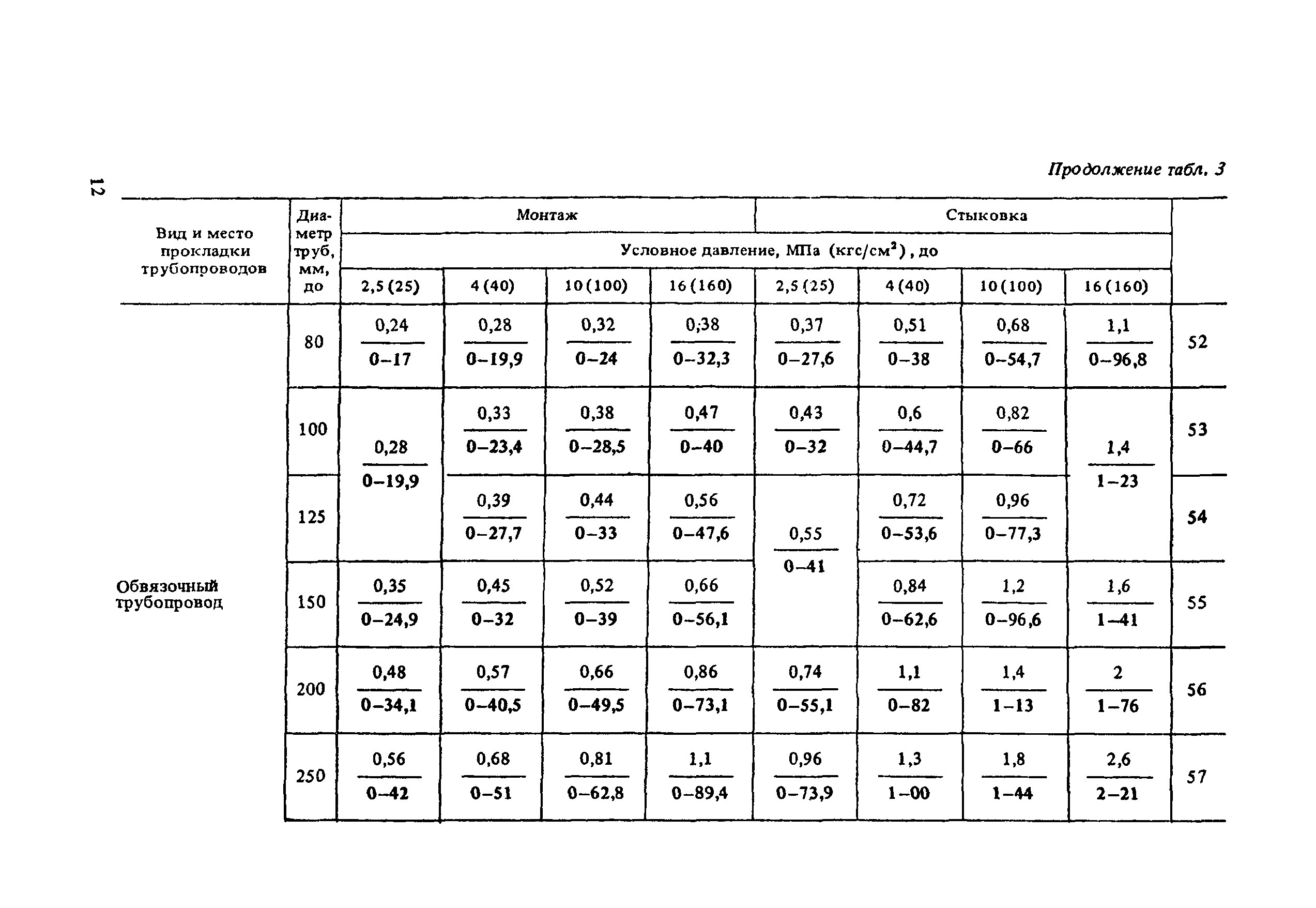 Как правило, сообщается об уровнях точности генотипа в диапазоне от 92 до 98% с небольшими различиями, наблюдаемыми между видами и типами популяций [9, 16, 19]. Преимущество использования данных повторного секвенирования таким образом заключается в том, что мы можем напрямую оценить точность данных GBS, полученных из разных конвейеров.
Как правило, сообщается об уровнях точности генотипа в диапазоне от 92 до 98% с небольшими различиями, наблюдаемыми между видами и типами популяций [9, 16, 19]. Преимущество использования данных повторного секвенирования таким образом заключается в том, что мы можем напрямую оценить точность данных GBS, полученных из разных конвейеров.
Еще одно важное соображение заключается в том, согласуются ли каталоги SNP, созданные с использованием разных конвейеров и разных технологий секвенирования.При использовании одной технологии секвенирования (Illumina) мы обнаружили, что около 80% или более SNP, вызываемых большинством конвейеров, также присутствовали в каталоге SNP, полученном из Fast-GBS. Таким образом, эти конвейеры в значительной степени согласуются с локусами, полиморфными в пределах данного набора зародышевой плазмы. Единственным исключением был TASSEL-GBS v1, так как только четверть SNP, представленных в результирующем каталоге, также присутствовала в наборе, полученном с помощью Fast-GBS. Вероятно, это связано с более короткими последовательностями (всего 64 п.н.) и большим количеством «ложных» SNP, поскольку этот конвейер оказался наименее точным из всех.При использовании одного и того же конвейера для анализа данных, полученных с помощью двух технологий секвенирования (Illumina и Ion Torrent), мы обычно обнаруживали, что перекрытие между каталогами SNP варьировалось примерно от 50 до 70%. Таким образом, выбор используемой технологии секвенирования привел к большей изменчивости в каталоге полученных SNP, чем выбор конвейера, используемого для одного набора прочтений. На первый взгляд может показаться, что это противоречит выводам, сделанным Mascher et al. (2013), которые обнаружили, что каталоги SNP, созданные с использованием двух конвейеров (TASSEL-GBS v1 и SAMtools), отличаются больше, чем каталоги, полученные с использованием разных технологий секвенирования (Illumina и Ion Torrent) [19].На наш взгляд, это скорее отражение ограничений TASSEL-GBS v1 (из-за коротких тегов).
Вероятно, это связано с более короткими последовательностями (всего 64 п.н.) и большим количеством «ложных» SNP, поскольку этот конвейер оказался наименее точным из всех.При использовании одного и того же конвейера для анализа данных, полученных с помощью двух технологий секвенирования (Illumina и Ion Torrent), мы обычно обнаруживали, что перекрытие между каталогами SNP варьировалось примерно от 50 до 70%. Таким образом, выбор используемой технологии секвенирования привел к большей изменчивости в каталоге полученных SNP, чем выбор конвейера, используемого для одного набора прочтений. На первый взгляд может показаться, что это противоречит выводам, сделанным Mascher et al. (2013), которые обнаружили, что каталоги SNP, созданные с использованием двух конвейеров (TASSEL-GBS v1 и SAMtools), отличаются больше, чем каталоги, полученные с использованием разных технологий секвенирования (Illumina и Ion Torrent) [19].На наш взгляд, это скорее отражение ограничений TASSEL-GBS v1 (из-за коротких тегов). Когда мы рассматриваем более широкий набор эталонных пайплайнов, они, как правило, обеспечивают очень хорошее перекрытие в непокрытых локусах SNP.
Когда мы рассматриваем более широкий набор эталонных пайплайнов, они, как правило, обеспечивают очень хорошее перекрытие в непокрытых локусах SNP.
Выводы, сделанные в этой работе, вероятно, распространяются на другие организмы, имеющие сходные геномные особенности (геном среднего размера, диплоидный). Можно ожидать, что виды, пережившие недавние события дупликации всего генома, будут представлять большую проблему, поскольку в таких случаях, вероятно, возрастет риск смешения аллелей в одном и том же локусе и паралогах.У видов, у которых такие события происходили в более отдаленном прошлом, у паралогов было больше возможностей расходиться, что облегчало правильное картирование прочтений.
Таким образом, невозможно разработать единый конвейер, одинаково подходящий для любой ситуации. Именно здесь для пользователей становится важным иметь возможность изменять различные параметры в процессе вызова SNP. К сожалению, не все пайплайны в этом отношении одинаково «прозрачны» и дают одинаковые возможности для изменений.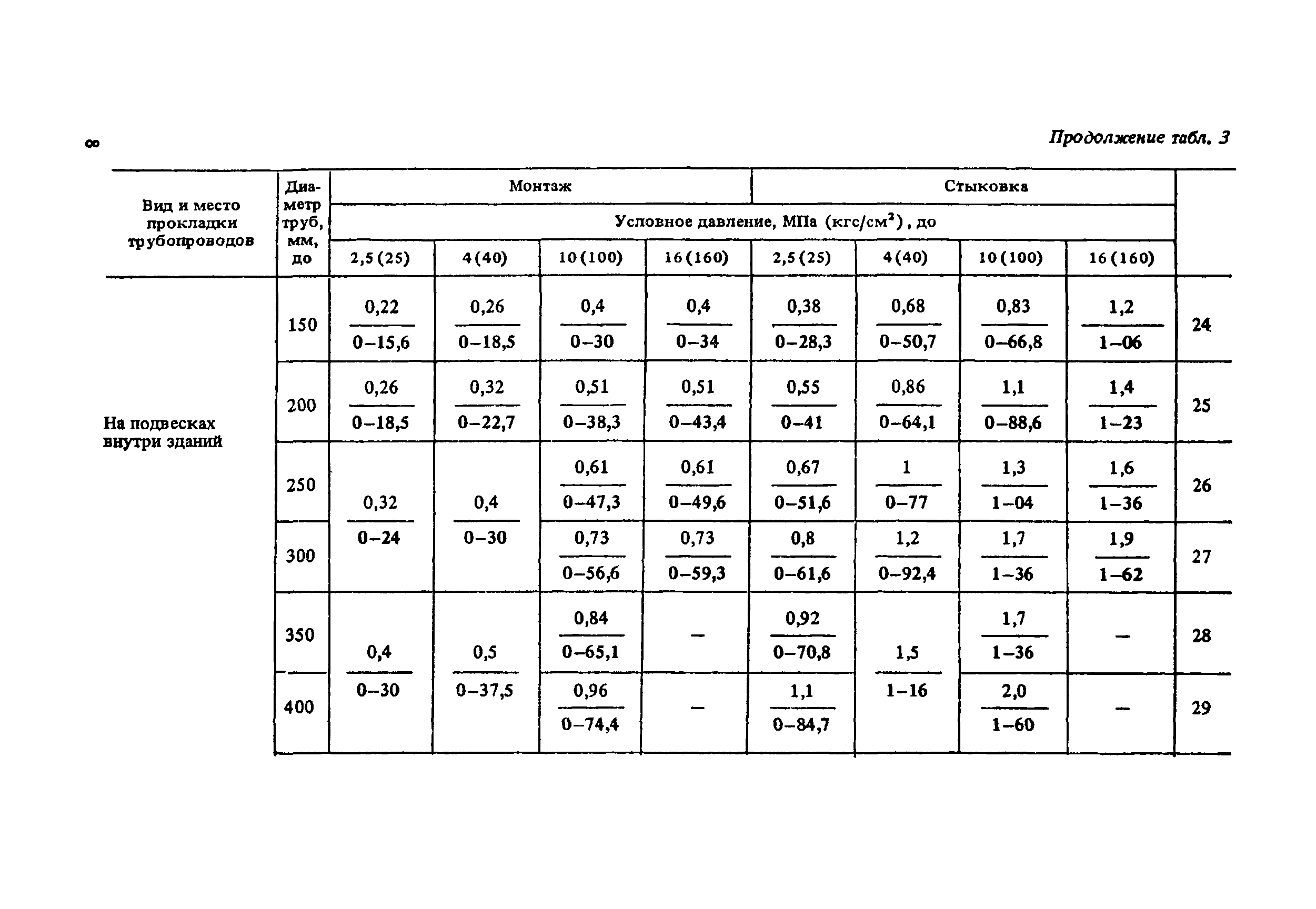 С одной стороны, UNEAK и TASSEL-GBS предлагают очень хорошую производительность, но полагаются на некоторые специально созданные инструменты или алгоритмы, которые пользователь не может легко изменить (например, для демультиплексирования и вариантного вызова). Кроме того, промежуточные файлы данных не всегда легкодоступны, что затрудняет исследование конкретных проблем. С другой стороны, IGST и Fast-GBS объединяют набор существующих инструментов, для которых пользователь может изменять параметры/параметры по своему желанию, а промежуточные файлы легко доступны.В этом спектре, на наш взгляд, Stacks предлагает промежуточный уровень прозрачности.
С одной стороны, UNEAK и TASSEL-GBS предлагают очень хорошую производительность, но полагаются на некоторые специально созданные инструменты или алгоритмы, которые пользователь не может легко изменить (например, для демультиплексирования и вариантного вызова). Кроме того, промежуточные файлы данных не всегда легкодоступны, что затрудняет исследование конкретных проблем. С другой стороны, IGST и Fast-GBS объединяют набор существующих инструментов, для которых пользователь может изменять параметры/параметры по своему желанию, а промежуточные файлы легко доступны.В этом спектре, на наш взгляд, Stacks предлагает промежуточный уровень прозрачности.
Наконец, хотя быстро приближается полногеномное секвенирование целых популяций, мы полагаем, что описанные здесь методы, вероятно, останутся бесценными в ближайшие годы в популяционной геномике, селекции, картировании и сборке референсных геномных последовательностей, особенно для не- модельные организмы.
Интегрированный конвейер добычи и использования SNP (ISMU) для данных секвенирования следующего поколения
PLoS One. 2014; 9(7): e101754.
2014; 9(7): e101754.
Сарвар Азам
1 Центр передового опыта в области геномики, Международный научно-исследовательский институт сельскохозяйственных культур для полузасушливых тропиков (ИКРИСАТ), Патанчеру, Индия,
Абхишек Ратор
1 Центр передового опыта в области геномики, Международный научно-исследовательский институт сельскохозяйственных культур для полузасушливых тропиков (ИКРИСАТ), Патанчеру, Индия,
Трушар М.Шах
1 Центр передового опыта в области геномики, Международный научно-исследовательский институт сельскохозяйственных культур для полузасушливых тропиков (ИКРИСАТ), Патанчеру, Индия,
Мохан Теллури
1 Центр передового опыта в области геномики, Международный научно-исследовательский институт сельскохозяйственных культур для полузасушливых тропиков (ИКРИСАТ), Патанчеру, Индия,
БхануПракаш Аминдала
1 Центр передового опыта в области геномики, Международный научно-исследовательский институт сельскохозяйственных культур для полузасушливых тропиков (ИКРИСАТ), Патанчеру, Индия,
Прадип Руперао
1 Центр передового опыта в области геномики, Международный научно-исследовательский институт сельскохозяйственных культур для полузасушливых тропиков (ИКРИСАТ), Патанчеру, Индия,
2 Школа сельского хозяйства и пищевых наук Университета Квинсленда, Брисбен, Австралия,
Мохан А.
 В. С. К. Катта
В. С. К. Катта1 Центр передового опыта в области геномики, Международный научно-исследовательский институт сельскохозяйственных культур для полузасушливых тропиков (ИКРИСАТ), Патанчеру, Индия,
Раджив К. Варшней
1 Центр передового опыта в области геномики, Международный научно-исследовательский институт сельскохозяйственных культур для полузасушливых тропиков (ИКРИСАТ), Патанчеру, Индия,
Манодж Прасад, редактор
1 Центр передового опыта в области геномики, Международный научно-исследовательский институт сельскохозяйственных культур для полузасушливых тропиков (ИКРИСАТ), Патанчеру, Индия,
2 Школа сельского хозяйства и пищевых наук Университета Квинсленда, Брисбен, Австралия,
Национальный институт исследований генома растений, Индия,
Конкурирующие интересы: Авторы заявили об отсутствии конкурирующих интересов.
Придумал и спроектировал эксперименты: РКВ.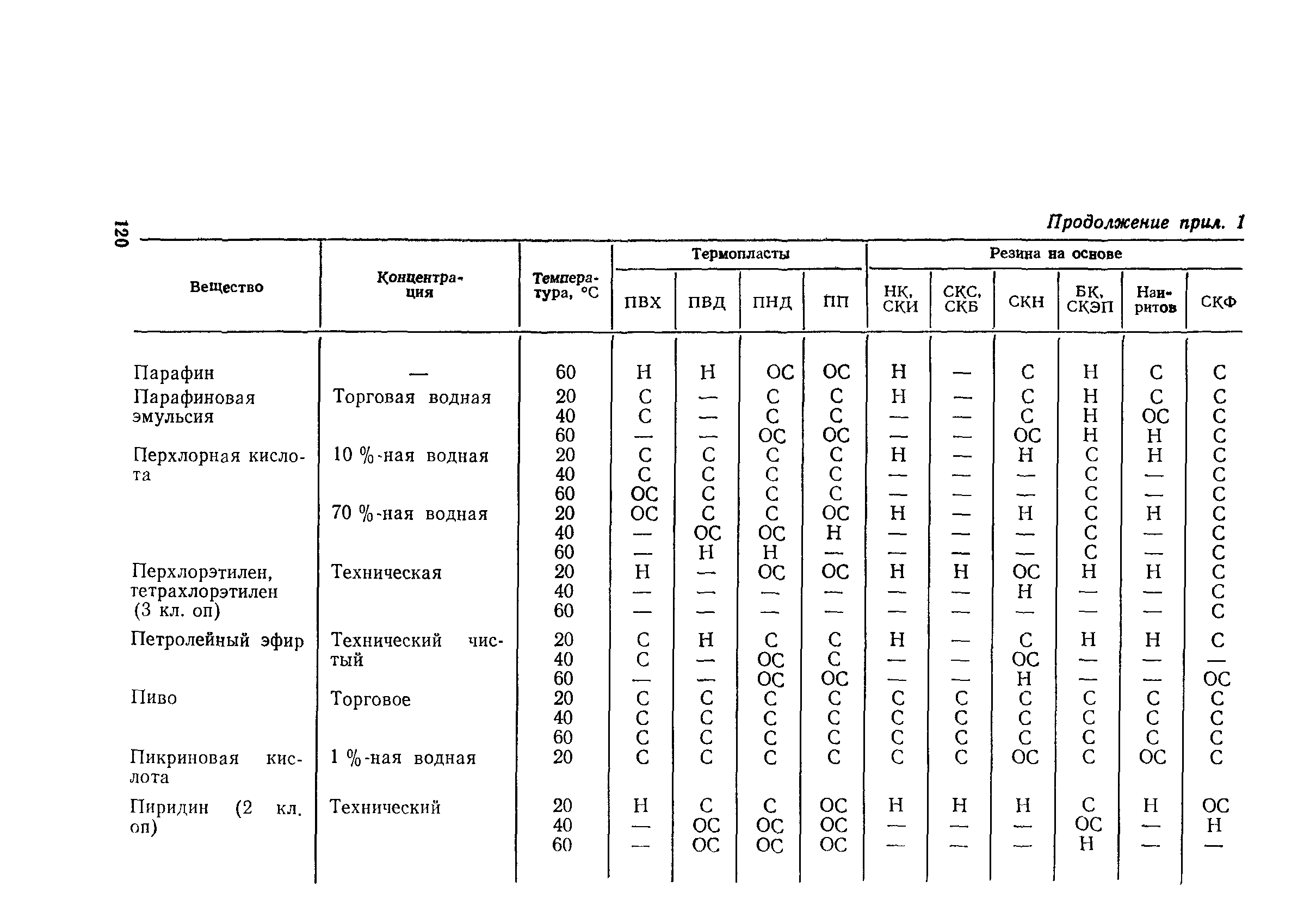 Выполнены опыты: СА АР ТС ПР МТ. Проанализированы данные: С.А. Предоставленные реагенты/материалы/инструменты для анализа: SA BPA PR MT. Написал статью: СА ТА МАВСКК РКВ.
Выполнены опыты: СА АР ТС ПР МТ. Проанализированы данные: С.А. Предоставленные реагенты/материалы/инструменты для анализа: SA BPA PR MT. Написал статью: СА ТА МАВСКК РКВ.
Поступила в редакцию 6 марта 2014 г .; Принято 11 июня 2014 г.
Это статья с открытым доступом, распространяемая в соответствии с лицензией Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии, что первоначальный автор и источник должным образом указаны.
Эта статья была процитирована другими статьями в PMC.Abstract
Конвейеры обнаружения однонуклеотидного полиморфизма (SNP) с открытым исходным кодом для данных секвенирования следующего поколения обычно требуют практических знаний интерфейса командной строки, огромных вычислительных ресурсов и опыта, что является сложной задачей для биологов. Кроме того, сгенерированная информация SNP не может быть легко использована для последующих процессов, таких как генотипирование.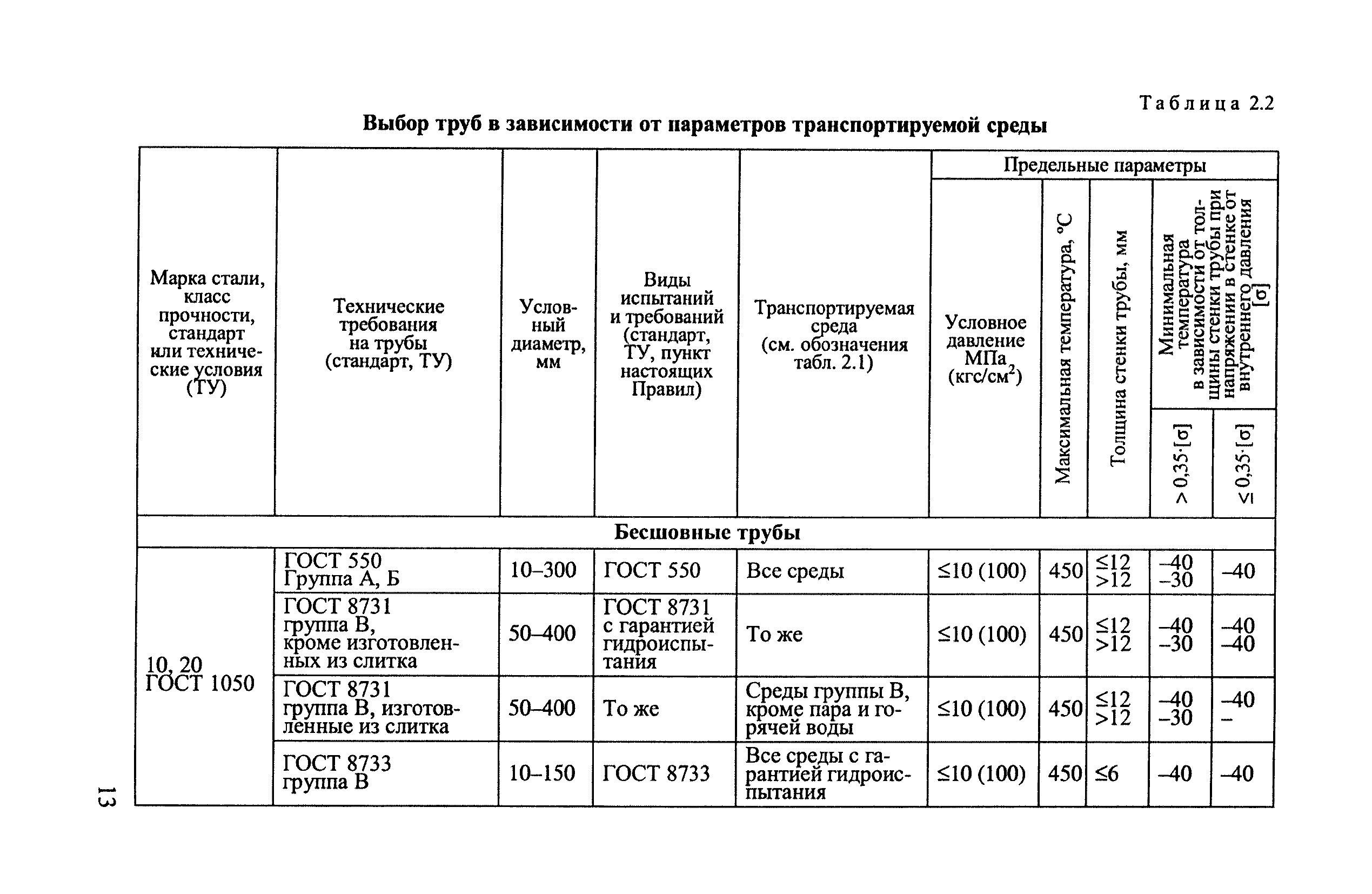 Таким образом, был разработан всеобъемлющий конвейер путем интеграции нескольких инструментов секвенирования следующего поколения (NGS) с открытым исходным кодом вместе с графическим пользовательским интерфейсом под названием «Интегрированный анализ и использование SNP» (ISMU) для обнаружения SNP и их использования при разработке анализов генотипирования.Конвейер включает такие функции, как предварительная обработка необработанных данных, интеграция инструментов выравнивания с открытым исходным кодом (Bowtie2, BWA, Maq, NovoAlign и SOAP2), методы прогнозирования SNP (SAMtools/SOAPsnp/CNS2snp и CbCC) и интерфейсы для разработки анализов генотипирования. Конвейер выводит список SNP высокого качества между всеми проанализированными парными комбинациями генотипов в дополнение к эталонному геному/последовательности. Инструменты визуализации (Tablet и Flapjack), интегрированные в конвейер, позволяют проверять выравнивание и ошибки, если таковые имеются.Конвейер также предоставляет показатель достоверности или значение информационного содержания полиморфизма с фланкирующими последовательностями для идентифицированных SNP в стандартном формате, необходимом для разработки анализов маркерного генотипирования (KASP и Golden Gate).
Таким образом, был разработан всеобъемлющий конвейер путем интеграции нескольких инструментов секвенирования следующего поколения (NGS) с открытым исходным кодом вместе с графическим пользовательским интерфейсом под названием «Интегрированный анализ и использование SNP» (ISMU) для обнаружения SNP и их использования при разработке анализов генотипирования.Конвейер включает такие функции, как предварительная обработка необработанных данных, интеграция инструментов выравнивания с открытым исходным кодом (Bowtie2, BWA, Maq, NovoAlign и SOAP2), методы прогнозирования SNP (SAMtools/SOAPsnp/CNS2snp и CbCC) и интерфейсы для разработки анализов генотипирования. Конвейер выводит список SNP высокого качества между всеми проанализированными парными комбинациями генотипов в дополнение к эталонному геному/последовательности. Инструменты визуализации (Tablet и Flapjack), интегрированные в конвейер, позволяют проверять выравнивание и ошибки, если таковые имеются.Конвейер также предоставляет показатель достоверности или значение информационного содержания полиморфизма с фланкирующими последовательностями для идентифицированных SNP в стандартном формате, необходимом для разработки анализов маркерного генотипирования (KASP и Golden Gate).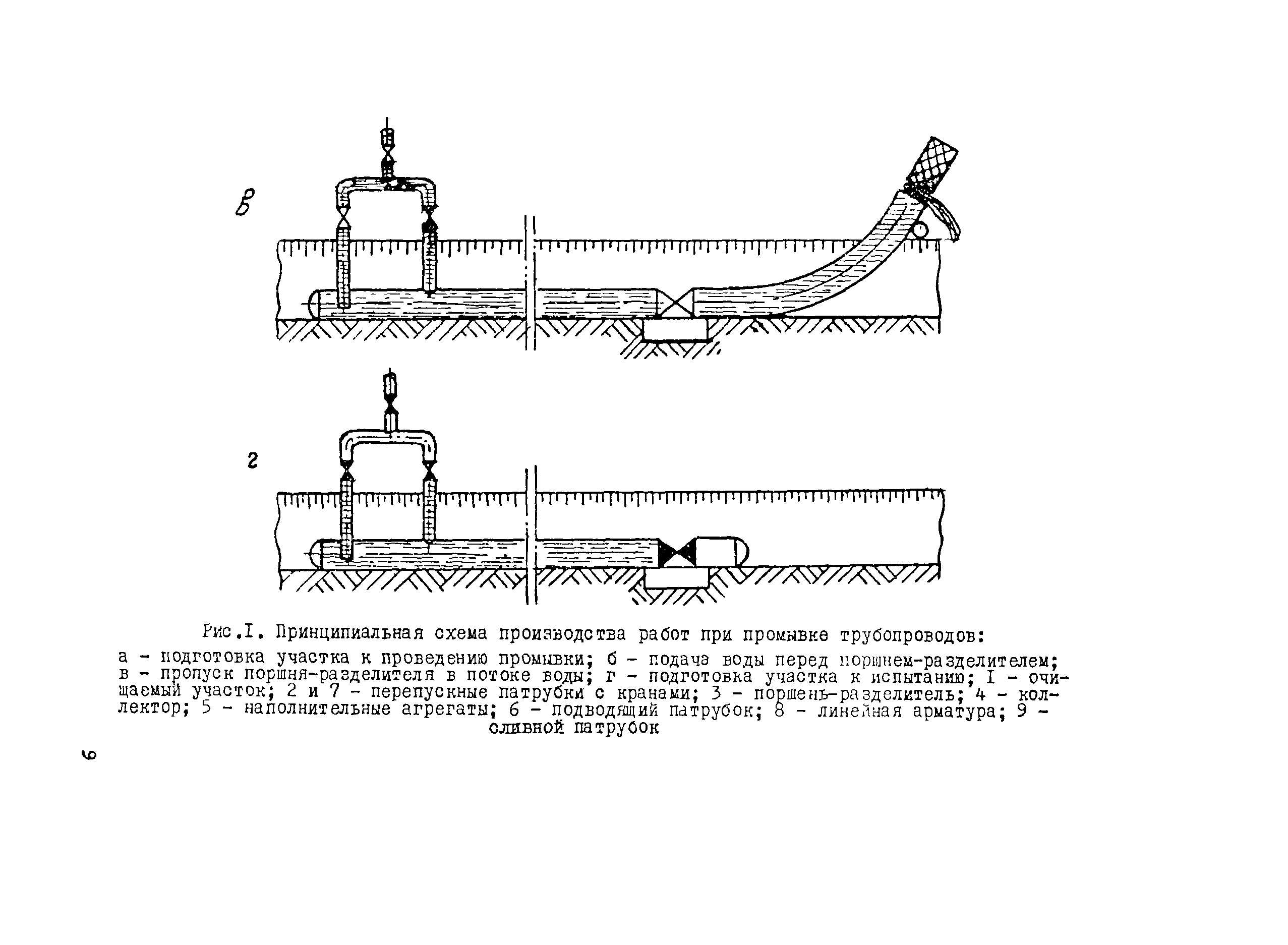 Конвейер позволяет пользователям обрабатывать ряд наборов данных NGS, таких как повторное секвенирование всего генома, секвенирование ДНК, связанное с сайтом рестрикции, и данные секвенирования транскриптома с высокой скоростью. Конвейер очень полезен для сообщества генетиков и селекционеров растений, не имеющих опыта в области вычислений, для обнаружения SNP и использования в геномных, генетических и селекционных исследованиях.Конвейер был распараллелен для обработки огромных наборов данных секвенирования следующего поколения. Он был разработан на языке Java и доступен по адресу http://hpc.icrisat.cgiar.org/ISMU как отдельное бесплатное программное обеспечение.
Конвейер позволяет пользователям обрабатывать ряд наборов данных NGS, таких как повторное секвенирование всего генома, секвенирование ДНК, связанное с сайтом рестрикции, и данные секвенирования транскриптома с высокой скоростью. Конвейер очень полезен для сообщества генетиков и селекционеров растений, не имеющих опыта в области вычислений, для обнаружения SNP и использования в геномных, генетических и селекционных исследованиях.Конвейер был распараллелен для обработки огромных наборов данных секвенирования следующего поколения. Он был разработан на языке Java и доступен по адресу http://hpc.icrisat.cgiar.org/ISMU как отдельное бесплатное программное обеспечение.
Введение
Технология секвенирования следующего поколения (NGS) изменила ландшафт исследований геномики, особенно геномики сельскохозяйственных культур, за последние несколько лет [1]–[4]. Illumina, платформы секвенирования SOLiD и Ion Torrent от Life Technologies использовались для быстрой идентификации SNP и других исследований маркеров в сельскохозяйственных культурах.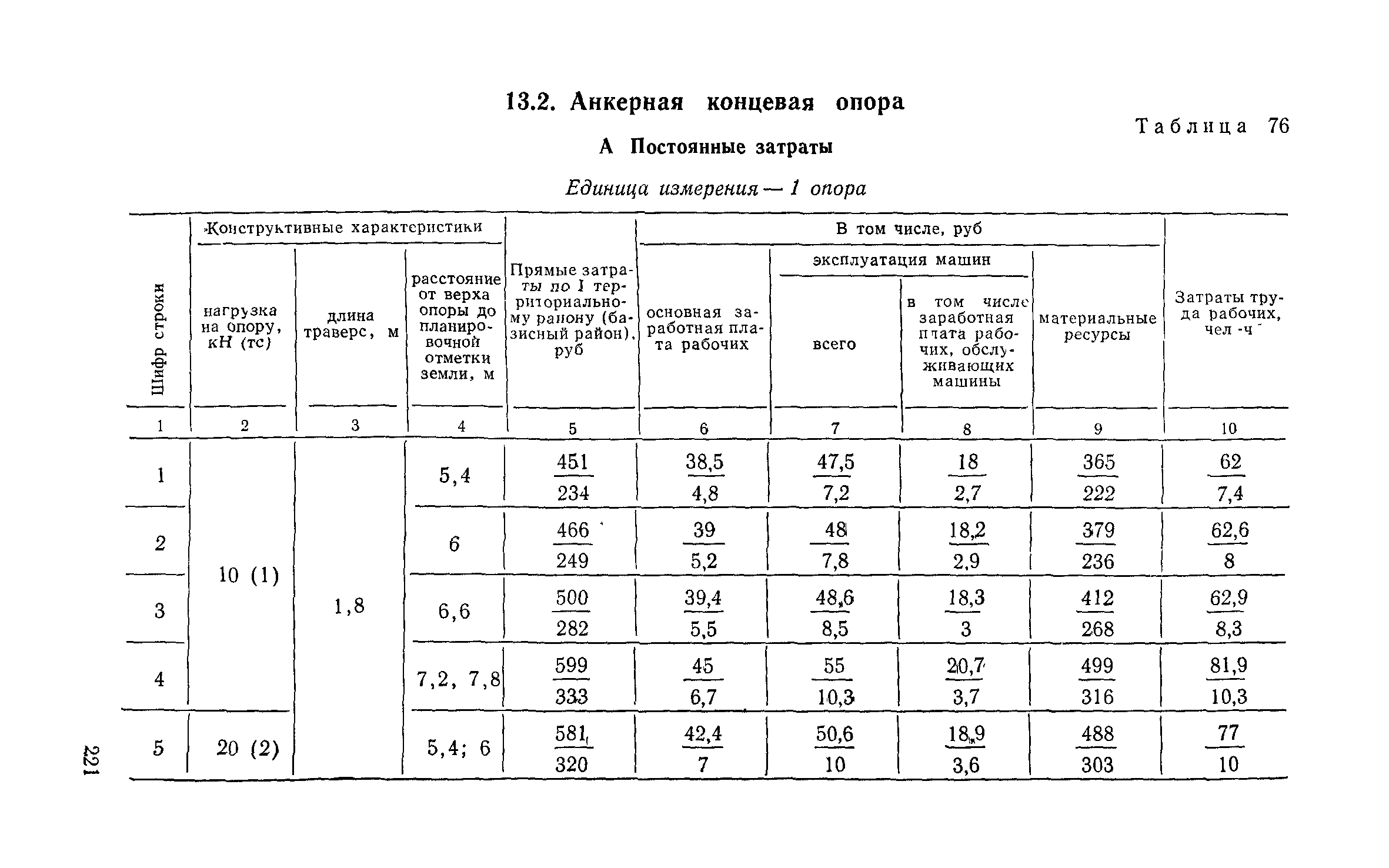 Эти платформы обычно производят короткие чтения размером 50–150 п.н., которые предпочтительны для идентификации SNP. Быстрый технологический прогресс в последнее время позволил технологиям NGS обеспечить значительно более высокую пропускную способность при меньших затратах. Однако стоимость экспериментов по секвенированию очень высока, особенно для видов с большими геномами или видов, для которых эталонный геном недоступен (сиротские культуры). В таких случаях секвенирование транскриптома из более чем одного генотипа является первым выбором для генетики растений и сообщества молекулярной селекции для расшифровки SNP.Преимущество этого подхода состоит в том, что идентифицированные SNP в основном расположены в однокопийных генах, что является необходимым условием для анализа SNP-маркеров. В различных моделях или основных видах сельскохозяйственных культур секвенирование транскриптома использовалось для обнаружения аллелей и анализа экспрессии генов [5]–[9]. Здесь ограниченное количество SNP, обнаруженных в генах, было связано с ограничениями отбора в кодирующих областях, что приводит к обнаружению только нескольких тысяч полезных маркеров.
Эти платформы обычно производят короткие чтения размером 50–150 п.н., которые предпочтительны для идентификации SNP. Быстрый технологический прогресс в последнее время позволил технологиям NGS обеспечить значительно более высокую пропускную способность при меньших затратах. Однако стоимость экспериментов по секвенированию очень высока, особенно для видов с большими геномами или видов, для которых эталонный геном недоступен (сиротские культуры). В таких случаях секвенирование транскриптома из более чем одного генотипа является первым выбором для генетики растений и сообщества молекулярной селекции для расшифровки SNP.Преимущество этого подхода состоит в том, что идентифицированные SNP в основном расположены в однокопийных генах, что является необходимым условием для анализа SNP-маркеров. В различных моделях или основных видах сельскохозяйственных культур секвенирование транскриптома использовалось для обнаружения аллелей и анализа экспрессии генов [5]–[9]. Здесь ограниченное количество SNP, обнаруженных в генах, было связано с ограничениями отбора в кодирующих областях, что приводит к обнаружению только нескольких тысяч полезных маркеров.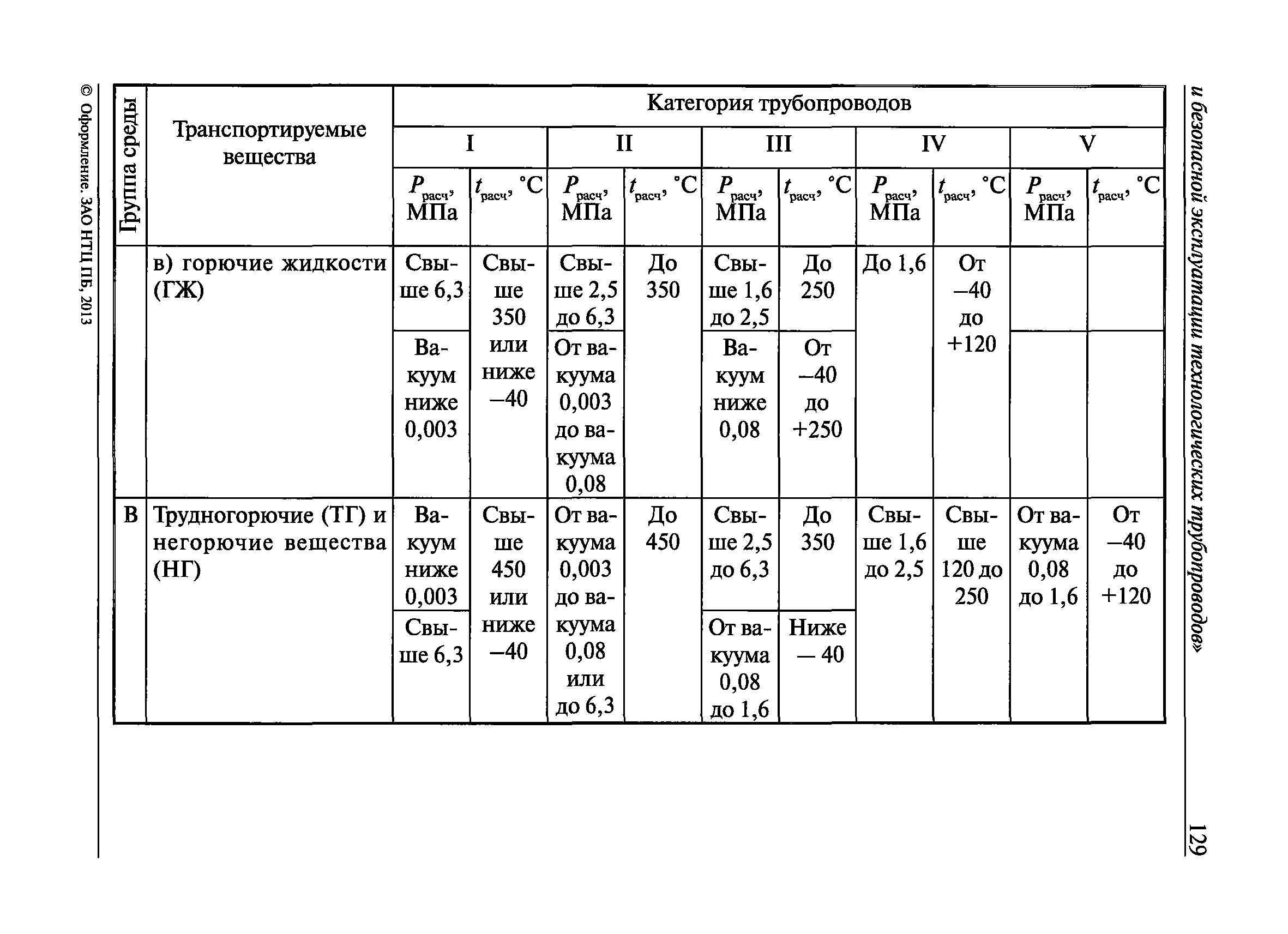 Для преодоления этого ограничения были разработаны альтернативные подходы.Эти подходы используют технологии NGS в сочетании с технологиями снижения сложности [10]. Эти технологии снижения сложности охватывают весь геном, не ограничиваясь областями, кодирующими белок. Следовательно, другие однокопийные последовательности также могут быть исследованы на наличие SNP. Технологии снижения сложности основаны, например, на селективном секвенировании фракции ДНК, полученной в результате расщепления чувствительными к метилированию рестрикционными ферментами [11]–[14], предварительной амплификации со специфическими комбинациями праймеров AFLP (амплифицированный полиморфизм длины фрагмента) [11]–[14]. 15] или использование технологии секвенирования RAD (Restriction-site-associated DNA) [16]–[18].По сравнению с подходом, основанным на транскриптоме, технологии уменьшения сложности имеют то преимущество, что они находят применение более или менее независимо от размера генома. Тем не менее, технология NGS недавно использовалась для секвенирования и повторного секвенирования всего генома с целью поиска большого количества SNP для построения карт гаплотипов, изучения внутривидового разнообразия и проведения полногеномных ассоциативных исследований (GWAS) для признаков.
Для преодоления этого ограничения были разработаны альтернативные подходы.Эти подходы используют технологии NGS в сочетании с технологиями снижения сложности [10]. Эти технологии снижения сложности охватывают весь геном, не ограничиваясь областями, кодирующими белок. Следовательно, другие однокопийные последовательности также могут быть исследованы на наличие SNP. Технологии снижения сложности основаны, например, на селективном секвенировании фракции ДНК, полученной в результате расщепления чувствительными к метилированию рестрикционными ферментами [11]–[14], предварительной амплификации со специфическими комбинациями праймеров AFLP (амплифицированный полиморфизм длины фрагмента) [11]–[14]. 15] или использование технологии секвенирования RAD (Restriction-site-associated DNA) [16]–[18].По сравнению с подходом, основанным на транскриптоме, технологии уменьшения сложности имеют то преимущество, что они находят применение более или менее независимо от размера генома. Тем не менее, технология NGS недавно использовалась для секвенирования и повторного секвенирования всего генома с целью поиска большого количества SNP для построения карт гаплотипов, изучения внутривидового разнообразия и проведения полногеномных ассоциативных исследований (GWAS) для признаков.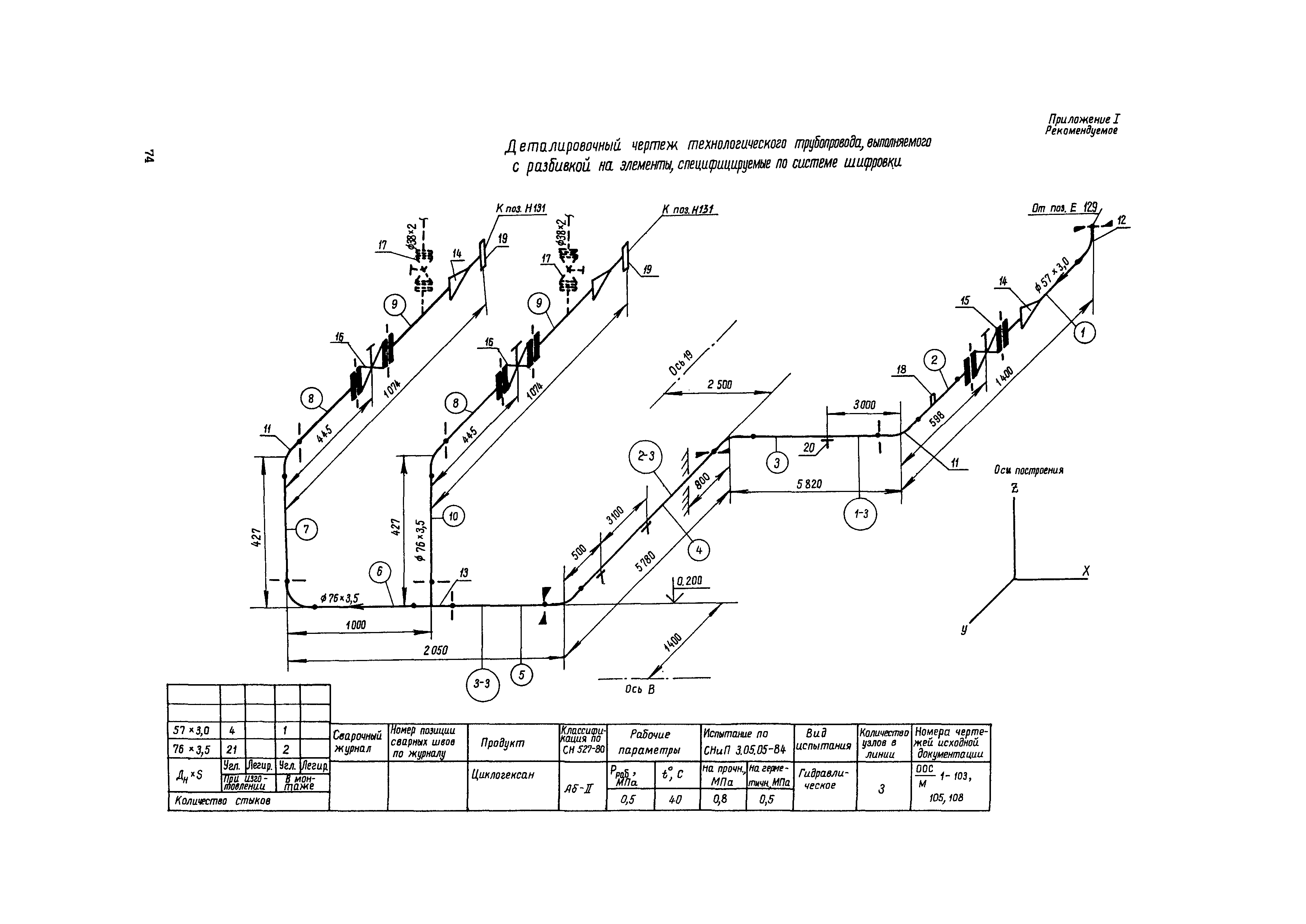 отображения [19]–[21].
отображения [19]–[21].
Идентифицированные SNP высокоэффективны только тогда, когда они распределены по всему геному и генотипированы для интересующей популяции или набора зародышевой плазмы.Как правило, обнаружение SNP с использованием NGS ограничено количеством проанализированных или секвенированных линий. Генотипирование большого количества SNP облегчает различные генетические анализы (например, филогенетический анализ, сверхплотное генетическое картирование и исследования ассоциации генотипа/фенотипа) и важные приложения (например, идентификацию сорта, селекцию с помощью маркеров). Наилучший подход к открытию SNP-маркеров для генотипирования — это сравнение полностью секвенированных геномов особей данного вида. Однако в случае, если полная эталонная последовательность недоступна, чтения могут быть собраны для получения мимической эталонной последовательности, и могут быть идентифицированы полногеномные SNP [22].В последние годы несколько таких сиротских культур были обогащены полной или частичной информацией о последовательности эталонного генома.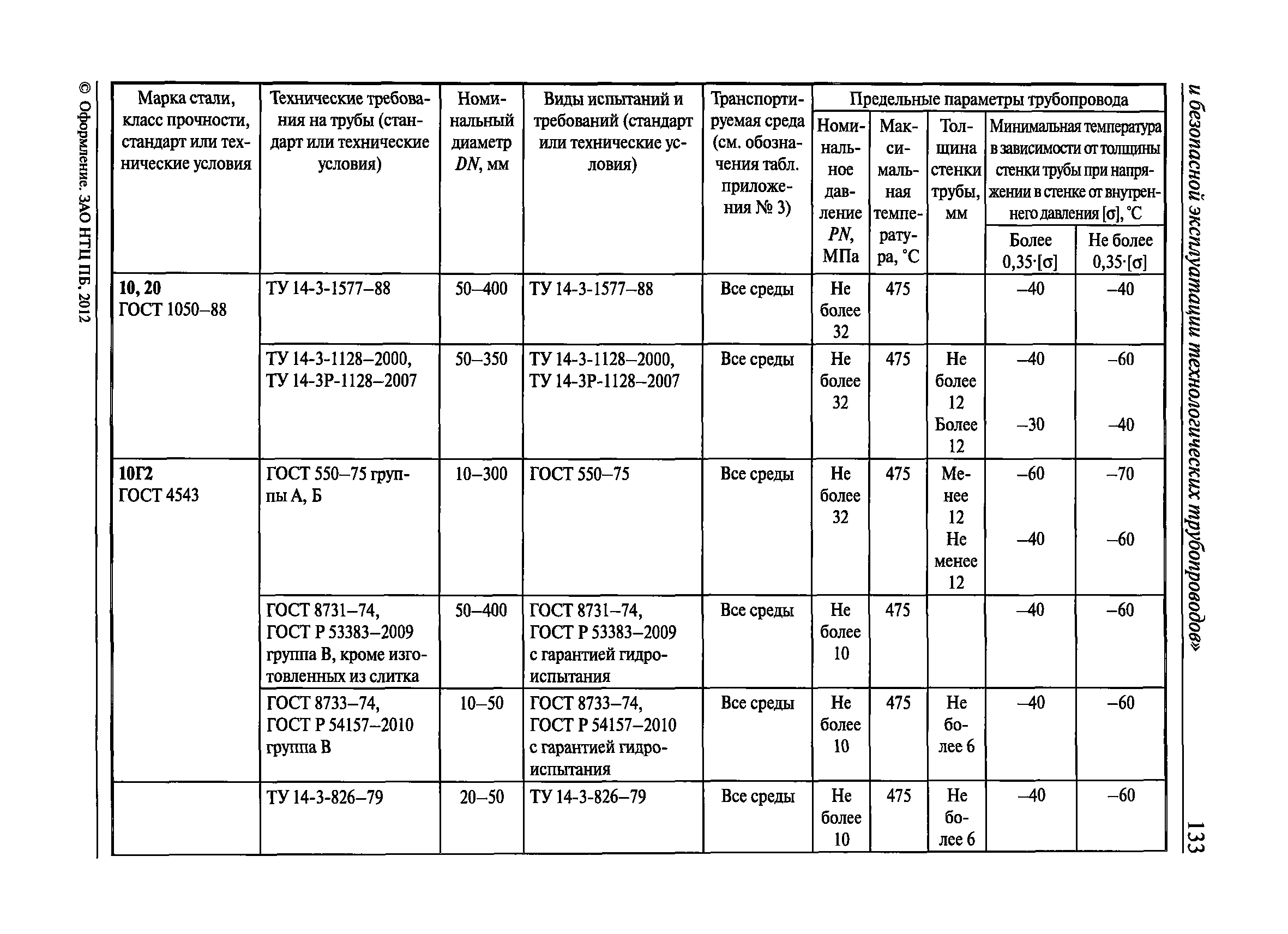 Сравнение данных повторного секвенирования с эталонной последовательностью генома дает большое количество SNP по всему геному и было описано для различных видов сельскохозяйственных культур, таких как нут [23], кукуруза [24], рис [25] и соя [26]. . Идентификация SNP позволяет использовать их в качестве маркеров в различных генетических анализах. Эти генетические анализы дополнительно требуют генотипирования маркеров SNP на полном наборе материала/популяции.Несмотря на то, что методы генотипирования ограничены стоимостью и временем подсчета SNP, наблюдается устойчивое развитие различных высокопроизводительных и недорогих автоматизированных платформ генотипирования. Эти платформы были в первую очередь разработаны для геномики человека, тем не менее, они были приняты и использованы для таких видов растений, как нут [23], [27], кукуруза [28], голубиный горох [29], рис [30] и пшеница [31]. . Они очень эффективны при обработке большого количества образцов и могут параллельно генотипировать от одного до миллиона SNP [32], [33].
Сравнение данных повторного секвенирования с эталонной последовательностью генома дает большое количество SNP по всему геному и было описано для различных видов сельскохозяйственных культур, таких как нут [23], кукуруза [24], рис [25] и соя [26]. . Идентификация SNP позволяет использовать их в качестве маркеров в различных генетических анализах. Эти генетические анализы дополнительно требуют генотипирования маркеров SNP на полном наборе материала/популяции.Несмотря на то, что методы генотипирования ограничены стоимостью и временем подсчета SNP, наблюдается устойчивое развитие различных высокопроизводительных и недорогих автоматизированных платформ генотипирования. Эти платформы были в первую очередь разработаны для геномики человека, тем не менее, они были приняты и использованы для таких видов растений, как нут [23], [27], кукуруза [28], голубиный горох [29], рис [30] и пшеница [31]. . Они очень эффективны при обработке большого количества образцов и могут параллельно генотипировать от одного до миллиона SNP [32], [33]. Некоторые популярные технологии, такие как TaqMan и SNPlex™ от Applied Biosystem/Life Technologies (Карлсбад, Калифорния, США), технологии на основе массивов, такие как технология GoldenGate и Infinium от Illumina (Сан-Диего, Калифорния, США), и технология на основе ПЦР, такая как KASP от LGC limited ( Теддингтон, Миддлсекс, Великобритания) являются наиболее востребованными вариантами исследований генотипирования для молекулярных селекционеров благодаря низкой стоимости, высокой производительности и автоматизированному рабочему процессу.
Некоторые популярные технологии, такие как TaqMan и SNPlex™ от Applied Biosystem/Life Technologies (Карлсбад, Калифорния, США), технологии на основе массивов, такие как технология GoldenGate и Infinium от Illumina (Сан-Диего, Калифорния, США), и технология на основе ПЦР, такая как KASP от LGC limited ( Теддингтон, Миддлсекс, Великобритания) являются наиболее востребованными вариантами исследований генотипирования для молекулярных селекционеров благодаря низкой стоимости, высокой производительности и автоматизированному рабочему процессу.
В общем, SNP можно использовать для обогащения геномных ресурсов вида в основном в два этапа: во-первых, это идентификация полиморфизмов последовательностей ДНК (обнаружение маркеров), а затем, во-вторых, анализ идентифицированных SNP-маркеров (генотипирование) по всему генотипу. сегрегация(и) популяции или набор зародышевой плазмы.В этом контексте стали доступны несколько конвейеров анализа данных, таких как ngs-backbone [34], SeqGene [35], Games [36], inGap [37] и GATK [38], которые поэтапно обрабатывают данные для идентификации SNP из данных NGS. . Эти конвейеры различаются по типу входных данных для анализа SNP. Например, некоторые конвейеры начинаются с необработанных данных, некоторые — с обработанных данных, а некоторые — с данных выравнивания. Эти конвейеры обычно предоставляют список SNP между эталонным генотипом и образцом. На самом деле низкая согласованность наблюдается среди SNP нескольких конвейеров вызовов SNP.В основном это связано с разнообразными интегрированными инструментами выравнивания и методами прогнозирования SNP. Кроме того, почти все конвейеры, доступные с открытым исходным кодом, основаны на командной строке и работают только на платформе Linux. Кроме того, идентифицированные SNP необходимо дополнительно обработать и отфильтровать, чтобы разработать тесты для их использования в качестве анализов маркерного генотипирования. Фактически, насколько нам известно, этап создания входного файла для отфильтрованных и высококачественных SNP для разработки анализа недоступен ни в одном конвейере. Таким образом, настоящее исследование было предпринято с общей целью разработать автоматизированный конвейер обнаружения SNP на основе графического пользовательского интерфейса (GUI) из данных NGS.
. Эти конвейеры различаются по типу входных данных для анализа SNP. Например, некоторые конвейеры начинаются с необработанных данных, некоторые — с обработанных данных, а некоторые — с данных выравнивания. Эти конвейеры обычно предоставляют список SNP между эталонным генотипом и образцом. На самом деле низкая согласованность наблюдается среди SNP нескольких конвейеров вызовов SNP.В основном это связано с разнообразными интегрированными инструментами выравнивания и методами прогнозирования SNP. Кроме того, почти все конвейеры, доступные с открытым исходным кодом, основаны на командной строке и работают только на платформе Linux. Кроме того, идентифицированные SNP необходимо дополнительно обработать и отфильтровать, чтобы разработать тесты для их использования в качестве анализов маркерного генотипирования. Фактически, насколько нам известно, этап создания входного файла для отфильтрованных и высококачественных SNP для разработки анализа недоступен ни в одном конвейере. Таким образом, настоящее исследование было предпринято с общей целью разработать автоматизированный конвейер обнаружения SNP на основе графического пользовательского интерфейса (GUI) из данных NGS. Мы назвали его интегрированным конвейером добычи и использования SNP (ISMU). Конвейер идентифицирует SNP из данных NGS образцов и извлекает информативные SNP для платформ генотипирования, таких как Illumina (BeadXpress, GoldenGate, Infinium) и KASP. ISMU поддерживает данные с одного конца (SE), а также с парными концами (PE) и перечисляет информативные SNP со значениями информационного содержания полиморфизма (PIC). Для облегчения анализа в небольших лабораториях с ограниченными вычислительными возможностями конвейер распространяется по запросу в виде образа виртуальной машины, а также на компакт-диске (CD).
Мы назвали его интегрированным конвейером добычи и использования SNP (ISMU). Конвейер идентифицирует SNP из данных NGS образцов и извлекает информативные SNP для платформ генотипирования, таких как Illumina (BeadXpress, GoldenGate, Infinium) и KASP. ISMU поддерживает данные с одного конца (SE), а также с парными концами (PE) и перечисляет информативные SNP со значениями информационного содержания полиморфизма (PIC). Для облегчения анализа в небольших лабораториях с ограниченными вычислительными возможностями конвейер распространяется по запросу в виде образа виртуальной машины, а также на компакт-диске (CD).
Results
ISMU представляет собой простой в использовании конвейер на основе графического интерфейса пользователя для обнаружения SNP, полезный для экспериментальных биологов, генетиков и специалистов по молекулярной селекции растений. Это позволяет исследователям анализировать данные NGS, сгенерированные Illumina, Life Technologies (SOLiD), или повторно анализировать уже депонированные данные из SRA (архив чтения последовательностей) NCBI. Он легко настраивается и объединяет популярные инструменты на основе командной строки для выравнивания и прогнозирования SNP с поддержкой данных как SE, так и PE в стандартном формате FASTQ [39].Он предназначен для работы на 64-битных настольных компьютерах. Поддержка многопоточности в ISMU позволяет пользователю анализировать большие объемы данных. Конвейер предоставляет высококачественные SNP, которые можно использовать для разработки тестов генотипирования для платформы KASP и Illumina. Все файлы результатов представляют собой плоские текстовые файлы, за исключением электронной таблицы, а именно «allele_data.xls». Конвейер дополнительно генерирует список INDEL, если для выравнивания использовался BWA/Bowie2.
Он легко настраивается и объединяет популярные инструменты на основе командной строки для выравнивания и прогнозирования SNP с поддержкой данных как SE, так и PE в стандартном формате FASTQ [39].Он предназначен для работы на 64-битных настольных компьютерах. Поддержка многопоточности в ISMU позволяет пользователю анализировать большие объемы данных. Конвейер предоставляет высококачественные SNP, которые можно использовать для разработки тестов генотипирования для платформы KASP и Illumina. Все файлы результатов представляют собой плоские текстовые файлы, за исключением электронной таблицы, а именно «allele_data.xls». Конвейер дополнительно генерирует список INDEL, если для выравнивания использовался BWA/Bowie2.
Особенности конвейера
Идентификация SNP — это многоэтапный процесс, включающий различные инструменты для предварительной обработки данных, сопоставления с эталоном и выявления вариаций с учетом фильтров.Рабочий процесс () можно в общих чертах разделить на шесть этапов следующим образом: (i) импорт данных, (ii) предварительная обработка данных, (iii) выравнивание последовательностей или картирование коротких прочтений на эталонном геноме, (iv) обнаружение SNP , (v) визуализация и (vi) генерация входных файлов анализа генотипирования.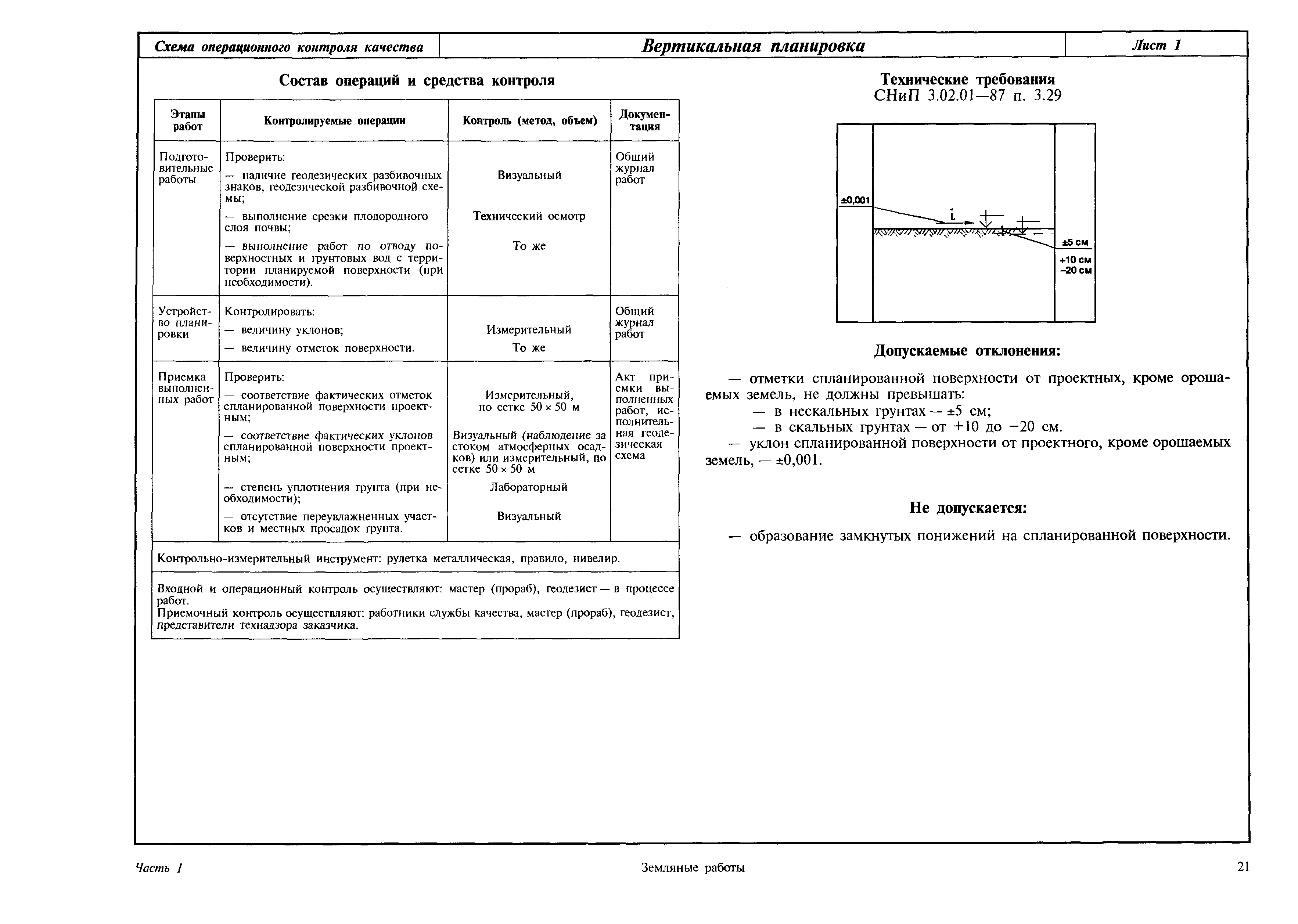 Все эти шаги были построены в автоматизированном конвейере ISMU для простоты использования. ISMU предлагает широкий выбор инструментов на каждом этапе с параметрами по умолчанию. Несколько вариантов позволяют сравнивать различные методологии (выравнивание и вызов SNP) и, таким образом, получать согласованный набор SNP для целевого генотипирования.
Все эти шаги были построены в автоматизированном конвейере ISMU для простоты использования. ISMU предлагает широкий выбор инструментов на каждом этапе с параметрами по умолчанию. Несколько вариантов позволяют сравнивать различные методологии (выравнивание и вызов SNP) и, таким образом, получать согласованный набор SNP для целевого генотипирования.
Рабочий процесс конвейера ISMU в основном разделен на три этапа: (A) импорт данных, предварительная обработка качества, (B) выравнивание последовательностей и обнаружение SNP, и (C) визуализация и создание входных файлов для анализа генотипа .
Импорт данных
Для конвейера требуются входные файлы в формате FASTQ [40]. Он принимает как парные, так и односторонние данные секвенирования. Однако существует несколько вариантов FASTQ (например, Illumina создает данные NGS в формате FASTQ с разными диапазонами показателей качества — Illumina 1.3, llumina 1.5 и Illumina 1.8), и было бы утомительно поддерживать их все.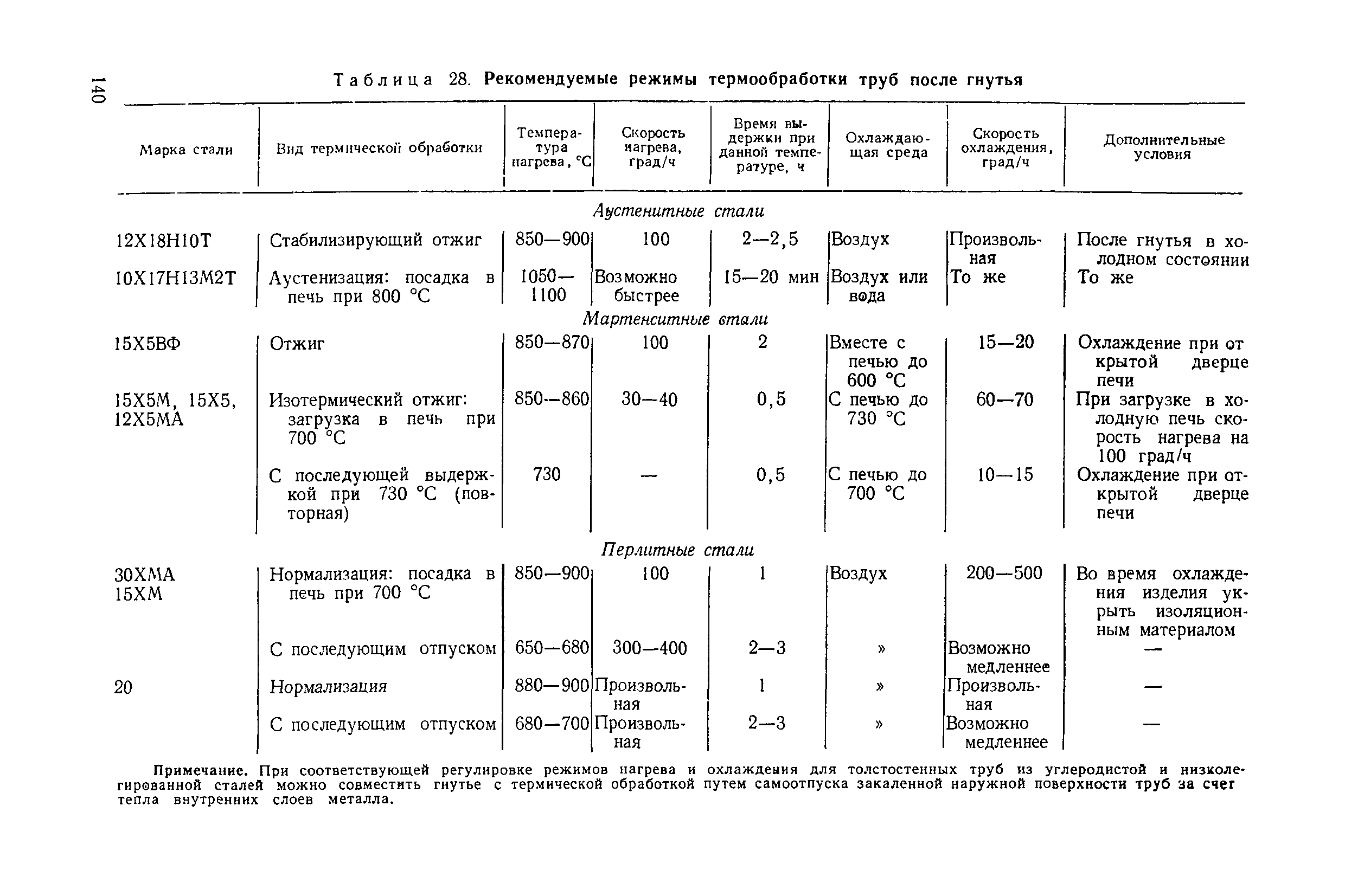 Чтобы обойти это несоответствие, конвейер принимает в качестве входных данных только файлы FASTQ (Sanger FASTQ), закодированные с помощью phred. Кроме того, требуется файл эталонной последовательности в формате FASTA, который будет использоваться в качестве шаблона для выравнивания входных наборов данных.
Чтобы обойти это несоответствие, конвейер принимает в качестве входных данных только файлы FASTQ (Sanger FASTQ), закодированные с помощью phred. Кроме того, требуется файл эталонной последовательности в формате FASTA, который будет использоваться в качестве шаблона для выравнивания входных наборов данных.
Предварительная обработка
Конвейер обрабатывает входные файлы FASTQ для оценки и создания обзора качества распределения набора данных. Предварительная обработка повышает качество входных данных за счет фильтрации и обрезки чтений.Он отфильтровывает чтения низкого качества, а затем обрезает области низкого качества в конце остаточных чтений. Этот этап предварительной обработки выполняется Perl-скриптом, который считывает входные данные и считает базы ридов низкокачественными, если показатель phred [40] меньше 20. Скрипт подсчитывает количество низкокачественных баз ридов в риде. и если процент таких баз превышает 30%, чтение помечается как низкое качество и отбрасывается. Затем независимо вычисляются средний показатель phred для каждой позиции чтения и средний показатель phred для всего набора данных.Эти оценки используются в качестве пороговых значений для обрезки низкокачественных оснований считываний либо с одного конца (5′ или 3′), либо с обоих концов, в зависимости от среднего качества оснований на концах. Порог установлен на 3 балла phred меньше, чем средний балл phred. Количество оснований, которые необходимо обрезать с концов, определяется динамически во время выполнения на основе внезапного изменения средних оценок phred, превышающих пороговое значение более чем на одну единицу оценки phred. Парные конечные данные далее обрабатываются путем отбрасывания всех чтений, не найденных парами.В результате создается FASTQ-файл с высоким качеством чтения для последующей обработки. Генерируется сводная статистика конвейера, описывающая качество необработанных данных, координаты обрезки и количество отфильтрованных чтений. Общее время обработки операций чтения на этом этапе зависит от количества входных данных и типа данных (SE или PE).
Затем независимо вычисляются средний показатель phred для каждой позиции чтения и средний показатель phred для всего набора данных.Эти оценки используются в качестве пороговых значений для обрезки низкокачественных оснований считываний либо с одного конца (5′ или 3′), либо с обоих концов, в зависимости от среднего качества оснований на концах. Порог установлен на 3 балла phred меньше, чем средний балл phred. Количество оснований, которые необходимо обрезать с концов, определяется динамически во время выполнения на основе внезапного изменения средних оценок phred, превышающих пороговое значение более чем на одну единицу оценки phred. Парные конечные данные далее обрабатываются путем отбрасывания всех чтений, не найденных парами.В результате создается FASTQ-файл с высоким качеством чтения для последующей обработки. Генерируется сводная статистика конвейера, описывающая качество необработанных данных, координаты обрезки и количество отфильтрованных чтений. Общее время обработки операций чтения на этом этапе зависит от количества входных данных и типа данных (SE или PE).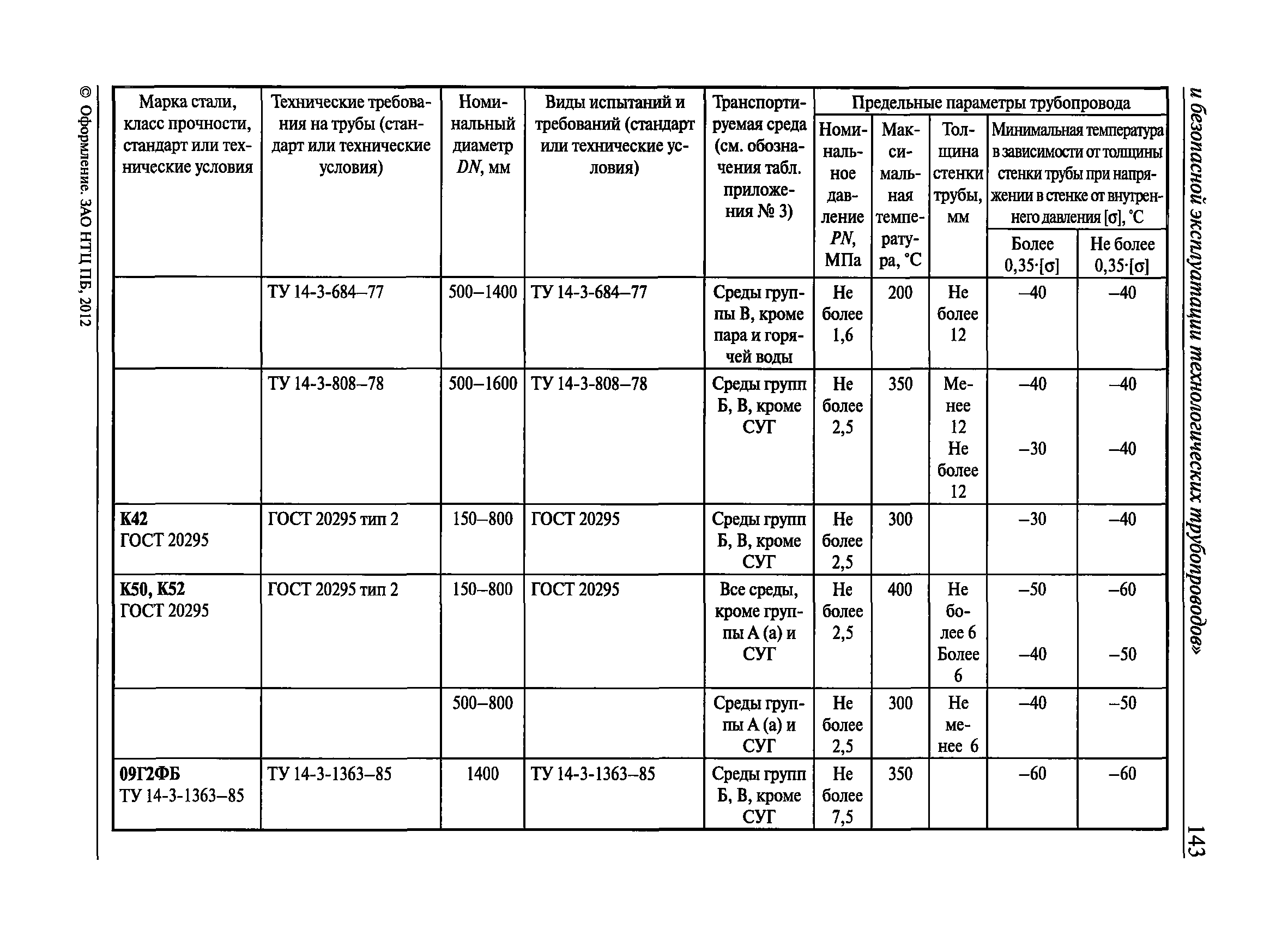 Этот этап предварительной обработки является необязательным.
Этот этап предварительной обработки является необязательным.
Выравнивание последовательности
Предварительно обработанные прочтения высокого качества сопоставляются с эталонной последовательностью.Инструменты выравнивания, а именно BWA [41], Maq [42], Bowtie2 [43], NovoAlign [44] и SOAP2 [45], были встроены в конвейер. Пользователь может выбрать любой из вышеупомянутых инструментов и предоставляет возможность изменить параметры по умолчанию. Значения параметров по умолчанию были оптимизированы и могут быть полезны для пользователей, не знакомых с инструментами. Этот шаг начинается с индексации эталонной последовательности для быстрого выравнивания, за которым следует сопоставление считываний, в результате чего получается результат выравнивания в формате SAM.По умолчанию выравнивания Maq и SOAP2 имеют формат MAP и были преобразованы в формат SAM с помощью Perl-скрипта «soap2sam.pl» [46]. Статистика выравнивания, сообщаемая инструментами, доступна в созданном файле журнала и сводном отчете конвейера. Статистика выравнивания предоставляет подробные сведения об объеме сопоставления и помогает пользователю понять, как параметры влияют на процесс сопоставления. В качестве альтернативы, файлы выравнивания формата SAM, созданные с помощью альтернативных инструментов сопоставления, могут использоваться для последующей обработки в ISMU.
Статистика выравнивания предоставляет подробные сведения об объеме сопоставления и помогает пользователю понять, как параметры влияют на процесс сопоставления. В качестве альтернативы, файлы выравнивания формата SAM, созданные с помощью альтернативных инструментов сопоставления, могут использоваться для последующей обработки в ISMU.
Идентификация SNP
Хорошее выравнивание с высокой глубиной считывания является важной предпосылкой для эффективного обнаружения SNP независимо от платформ секвенирования с данными NGS. Конвейер предоставляет два разных метода вызова SNP, а именно: (i) SAMtools [47] и (ii) согласованный вызов на основе покрытия (CbCC) [48]. Если выбран метод SAMtools, файлы формата SAM преобразуются в формат BAM, а bcftools используется для вызова вариаций (между эталоном и образцами) в формате VCF [49].Конвейер использует эту информацию, чтобы найти достоверный полиморфизм между образцами, а также между эталоном и образцами. SNP SAMtools дополнительно подтверждаются применением пользовательского фильтра следующим образом. Получается стек оснований в данной позиции SNP, которые удовлетворяют порогу качества phred >20. Частота результирующих оснований используется для достижения согласованного основания. Эта консенсусная база может быть основной или неоднозначной базой, представляющей гетерозиготность.Эти основания затем сравниваются между генотипами, чтобы вывести достоверные SNP. Результирующие SNP сообщаются с информацией о частоте по основному вызываемому основанию и их глубине чтения. В случае, если выравнивание формата SAM недоступно по умолчанию, например, при использовании выравнивателей Maq и SOAP, вместо SAMtools используются соответственно SOAPsnp [50] и Maq («cns2snp.pl»).
Получается стек оснований в данной позиции SNP, которые удовлетворяют порогу качества phred >20. Частота результирующих оснований используется для достижения согласованного основания. Эта консенсусная база может быть основной или неоднозначной базой, представляющей гетерозиготность.Эти основания затем сравниваются между генотипами, чтобы вывести достоверные SNP. Результирующие SNP сообщаются с информацией о частоте по основному вызываемому основанию и их глубине чтения. В случае, если выравнивание формата SAM недоступно по умолчанию, например, при использовании выравнивателей Maq и SOAP, вместо SAMtools используются соответственно SOAPsnp [50] и Maq («cns2snp.pl»).
В методе CbCC конвейер считывает файлы сэмплов SAM и извлекает информацию о наложениях во всех позициях. Эта информация была использована для расчета и сравнения консенсусного основания или консенсусного аллеля для каждого образца в каждом соответствующем положении ссылки.Если консенсусная база различается между образцами в этой позиции, то это указывается как SNP. Конвейер вычисляет частоту аллеля в каждой позиции выровненной последовательности. Если частота этого аллеля превышает порог F мажор , равный 0,66, он считается основным/консенсусным основанием [48].
Конвейер вычисляет частоту аллеля в каждой позиции выровненной последовательности. Если частота этого аллеля превышает порог F мажор , равный 0,66, он считается основным/консенсусным основанием [48].
Наконец, сообщаются SNP между образцами (попарно), а также относительно эталонной последовательности. Кроме того, полный неизбыточный набор SNP во всех образцах, а также в эталоне также сообщается как матрица генотипирования SNP.Все заявленные SNP описываются с указанием их положения в эталонной последовательности, основных оснований и соответствующего отношения достоверности в каждом из предоставленных образцов. Эти статистические данные также обеспечивают меру достоверности при выборе SNP для разработки тестов генотипирования.
Визуализация
Хотя этот шаг необязателен, визуализация выравнивания и распределения SNP, идентифицированных в образцах, поможет выявить любые возможные ошибки секвенирования или артефакты. Конвейер интегрирован с двумя инструментами визуализации, а именно Tablet [51] и Flapjack [52]. Планшет используется для визуализации выравнивания прочтений на эталонной последовательности и наблюдения за вызываемыми аллелями в положениях SNP. Он принимает информацию SNP в формате gff3 для прямого перехода к позициям SNP на дисплее выравнивания [53]. У пользователя есть возможность выбрать набор позиций SNP (, т. е. , SNP между любыми двумя образцами) из раскрывающегося списка в окне результатов для просмотра на планшете. Flapjack — это еще одно программное обеспечение для графического генотипирования на основе Java, которое можно использовать для визуализации SNP или распределения аллелей на каждой хромосоме среди образцов.Гаплотип можно просмотреть, и пользователь также может сгруппировать, сгруппировать или отсортировать генотипы/линии сходства с другими линиями.
Планшет используется для визуализации выравнивания прочтений на эталонной последовательности и наблюдения за вызываемыми аллелями в положениях SNP. Он принимает информацию SNP в формате gff3 для прямого перехода к позициям SNP на дисплее выравнивания [53]. У пользователя есть возможность выбрать набор позиций SNP (, т. е. , SNP между любыми двумя образцами) из раскрывающегося списка в окне результатов для просмотра на планшете. Flapjack — это еще одно программное обеспечение для графического генотипирования на основе Java, которое можно использовать для визуализации SNP или распределения аллелей на каждой хромосоме среди образцов.Гаплотип можно просмотреть, и пользователь также может сгруппировать, сгруппировать или отсортировать генотипы/линии сходства с другими линиями.
Входной файл для анализа генотипирования
На этом этапе конвейера создается входной файл для разработки методов анализа генотипа. Конвейер проверяет различные критерии выбора SNP, подходящих для генотипирования. Во-первых, SNP должен иметь полные фланкирующие последовательности с обеих сторон. Во-вторых, фланкирующие последовательности не должны содержать двусмысленных оснований типа «N», «K», «R».В-третьих, фланкирующие последовательности не должны содержать никаких других SNP. SNP, удовлетворяющие этим критериям, предпочтительны для анализа генотипирования (однако длина фланкирующей последовательности различается для платформ генотипирования KASP и Illumina). Все такие потенциальные SNP фильтруются, чтобы подготовить предварительный входной файл для разработки теста генотипирования. Менее информативные SNP можно исключить либо при визуальном осмотре, либо на основании значения PIC, чтобы в дальнейшем использовать высокоинформативные SNP в экспериментах по генотипированию.
Во-первых, SNP должен иметь полные фланкирующие последовательности с обеих сторон. Во-вторых, фланкирующие последовательности не должны содержать двусмысленных оснований типа «N», «K», «R».В-третьих, фланкирующие последовательности не должны содержать никаких других SNP. SNP, удовлетворяющие этим критериям, предпочтительны для анализа генотипирования (однако длина фланкирующей последовательности различается для платформ генотипирования KASP и Illumina). Все такие потенциальные SNP фильтруются, чтобы подготовить предварительный входной файл для разработки теста генотипирования. Менее информативные SNP можно исключить либо при визуальном осмотре, либо на основании значения PIC, чтобы в дальнейшем использовать высокоинформативные SNP в экспериментах по генотипированию.
Выходные данные конвейера
Выходные данные, созданные конвейером, включают сводный отчет, информацию SNP для попарной комбинации образцов среди образцов, входные файлы для генотипирования KASP и предварительный входной файл для оценки ADT для генотипирования Illumina. Все файлы находятся в текстовом файле, разделенном табуляцией, а также создается электронная таблица с именем файла «allele_data.xls». Помимо файлов журнала и vcf, создаются многие другие промежуточные файлы, такие как файлы SAM и BAM для каждого образца, а также отфильтрованные высококачественные наборы данных FASTQ, к которым можно получить доступ в автономном ISMU.Входные файлы также могут быть визуализированы.
Все файлы находятся в текстовом файле, разделенном табуляцией, а также создается электронная таблица с именем файла «allele_data.xls». Помимо файлов журнала и vcf, создаются многие другие промежуточные файлы, такие как файлы SAM и BAM для каждого образца, а также отфильтрованные высококачественные наборы данных FASTQ, к которым можно получить доступ в автономном ISMU.Входные файлы также могут быть визуализированы.
Оценка конвейера
Производительность, точность и скорость конвейера оценивалась с использованием двух наборов геномных и одного транскриптомного набора данных.
Предварительная обработка и выравнивание
Набор геномных данных включал данные повторного секвенирования всего генома (WGRS) четырех генотипов нута (Pistol, Slasher, Hat Trick и Genesis 90) и данные секвенирования RAD десяти генотипов нута (ICCV 03107, ICC 4918, ICC 4930, ICC 4958, ICC 5270, ICC 05530, ICC 5810, ICC 5912, ICC 6263 и ICC 8261).Набор транскриптомных данных включал данные RNAseq для двух генотипов арахиса (HuaU12 и HuaU606).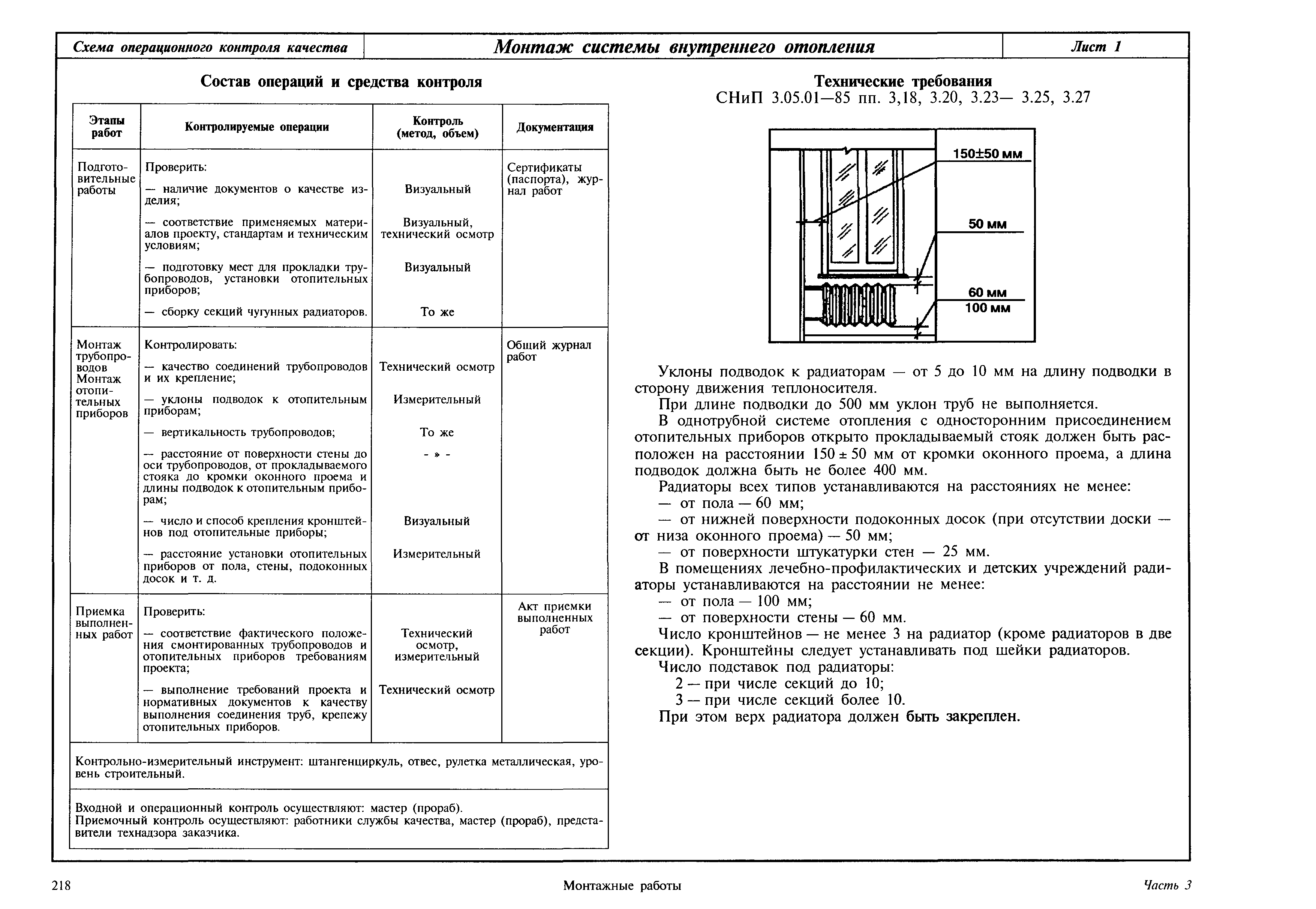 После предварительной обработки количество прочтений, соответствующих критериям фильтрации, в наборах данных WGRS, RAD и RNAseq составило 97,11% (125,07 млн), 95,39% (74,22 млн) и 98,18% (6,73 млн) соответственно. Наборы данных WGRS и RAD были сопоставлены с геномом нута, а набор данных RNAseq был сопоставлен с уникальными генами арахиса. В среднем 92,68% (), 90,5% () и 80,61% () прочтений сопоставлены с эталонными последовательностями из наборов данных WGRS, RAD и RNAseq соответственно.
После предварительной обработки количество прочтений, соответствующих критериям фильтрации, в наборах данных WGRS, RAD и RNAseq составило 97,11% (125,07 млн), 95,39% (74,22 млн) и 98,18% (6,73 млн) соответственно. Наборы данных WGRS и RAD были сопоставлены с геномом нута, а набор данных RNAseq был сопоставлен с уникальными генами арахиса. В среднем 92,68% (), 90,5% () и 80,61% () прочтений сопоставлены с эталонными последовательностями из наборов данных WGRS, RAD и RNAseq соответственно.
Таблица 1
Подробная информация о наборе данных повторного секвенирования всего генома (WGRS), использованном для оценки конвейера.
Длина считывания| Название генотипа | Тип | Общее количество необработанных чтений (PE) | Длина считывания (bp) | Общее количество отфильтрованных чтений (PE) | Выравнивание (%) | Количество SNP по сравнению с эталонным геномом | |||
| Пистолет | дези | 33 467 106 | 101 | 32 193 245 | 86/87 | 93. 03 03 | 317 991 | ||
| Хет-трик | дези | 39872 | 39 021614 | 101 | 31 12672 | 31 126432 | 86/87 | 92.53 | 156 255 |
| Slasher | дези | 35872 | 31 093,427 | 101 | 301 | 30 286 838 | 86/88 | 92.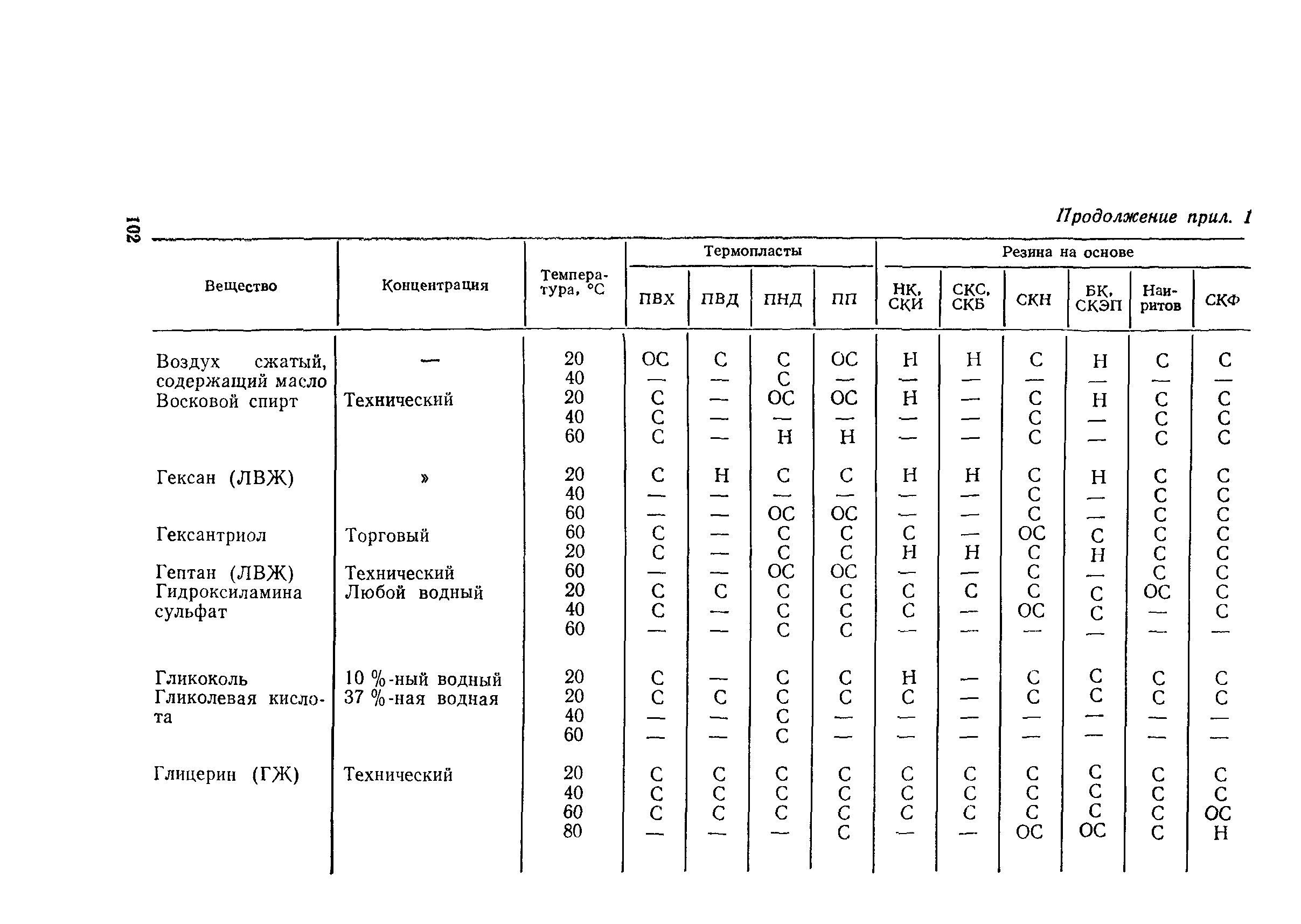 51 51 | 351 844 |
| Genesis | Кабули | 32 210 496 | 101 | 31 467 878 | 86/88 | 92.66 | 253 472 |
Таблица 2
Набор данных последовательности ДНК, связанной с сайтом рестрикции (RAD), используемый для оценки трубопровода.
| Название генотипа | Общее количество читаемых (SE) | Длина чтения (BP) | Общее количество фильтрованных читается | Длина чтения (BP) | выравнивание (%) | |
| ICCV 03107 | 2 360 400 | 100 | 2 250 687 | 78 | 91. 27 27 | |
| МТП 4918 | 5761446 | 100 | 5486801 | 78 | 89,82 | |
| МТП 4930 | 10595164 | 100 | 10103218 | 78 | 89,90 | |
| МТП 4958 | 10 874 599 | 100 | 10 400166 | 79 | 99 | 91.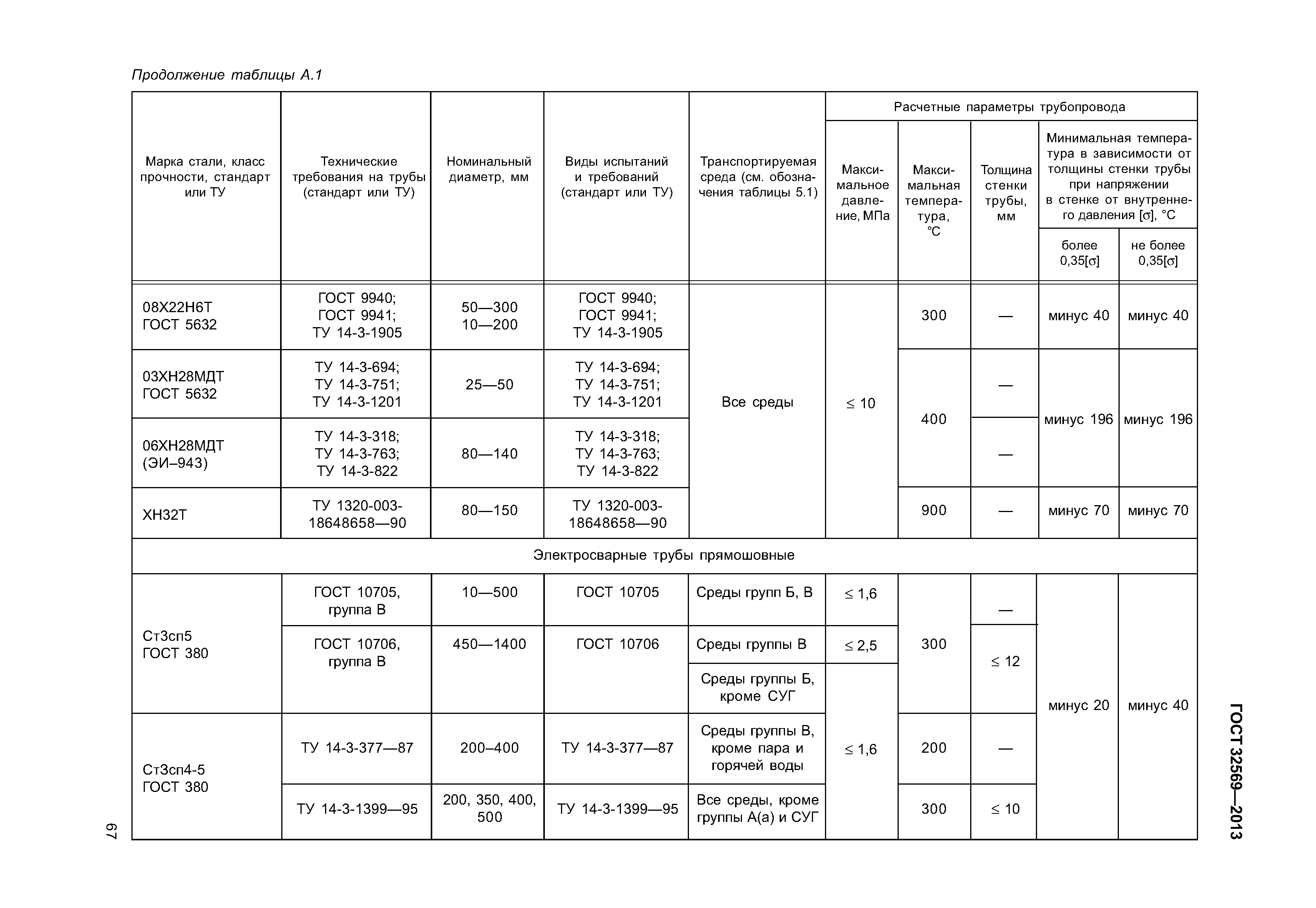 83 83 |
| ICC 5270 | 8,198 607 | 100 | 7 790 524 | 78 | 90.38 | |
| ICCV 05530 | 8011084 | 100 | 7611453 | 78 | 89,96 | |
| МТП 5810 | 8587698 | 100 | 8213783 | 79 | 90,68 | |
| МТП 5912 | 5422669 | 100 | 5183888 79 | |||
| 6263 | 8167648 | 100 | 7808763 79 | 90.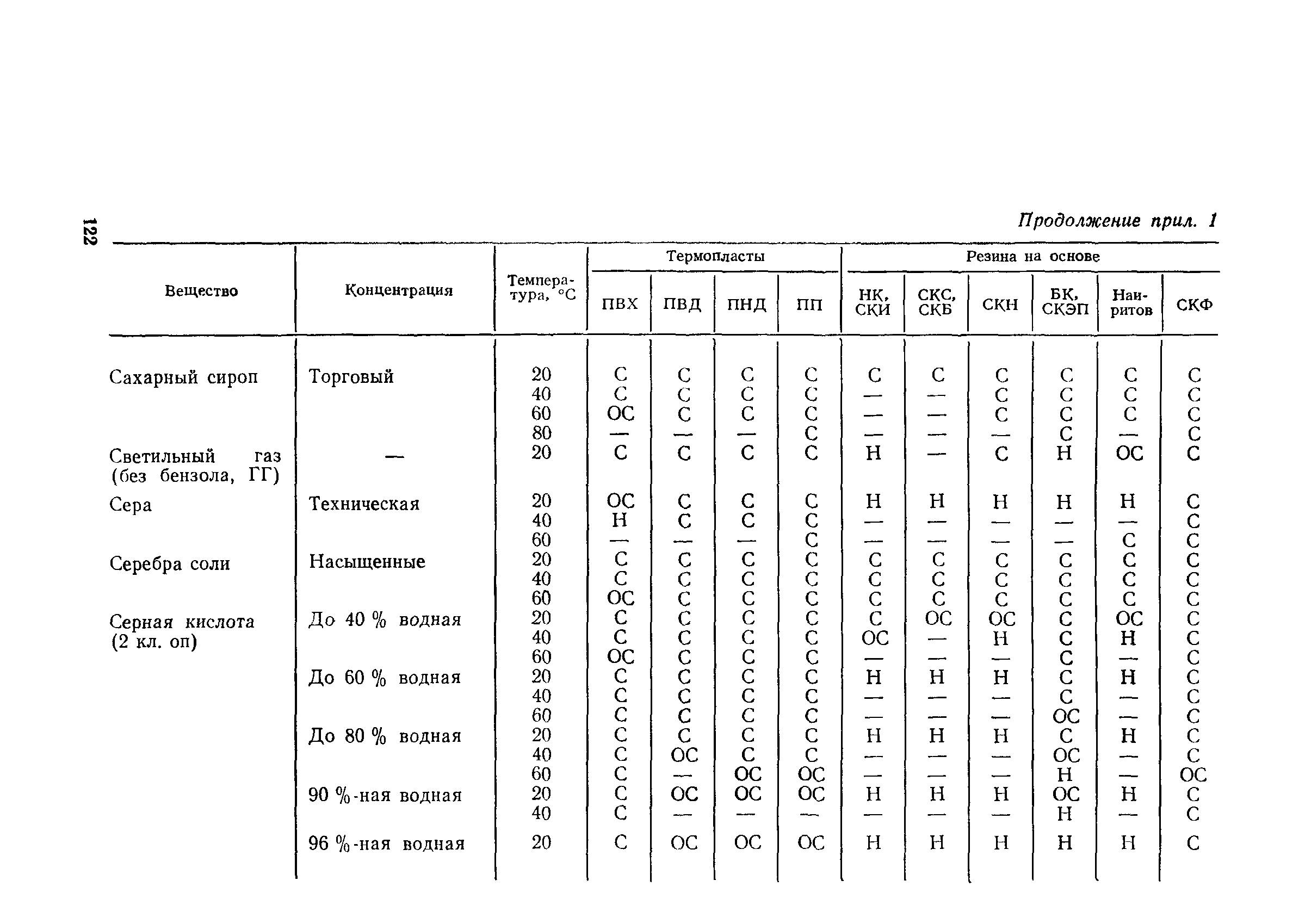 89 89 | ||
| ICC 8261 | 6,245,558 | 5 943, 309 | 5 943 309 | 81 | 88.94 | 88.94 |
Таблица 3
RNASEQ DataSet используются для оценки трубопровода.
| Название генотипа | RAW | отфильтрованные данные | выравниваются данные | выравнивание (%) | SNP со ссылкой | |||
| Общее количество читаемых (PE) | Длина чтения (BP) | Количество читаемых (PE) | 3 | |||||
| HUAU1 | 6,857,839 | 90/90 | 90/90 | 72/74 | 3 82. 51 51 | 41225 | ||
| HuaU606 6771173 | 90/90 6649229 | 72/74 78,71 | 44984 | |||||
В общей сложности 579,813 ОНП были определены на основе данных WGRS () против ссылки. Подмножество (62 291) этих SNP в каждом генотипе показало полиморфизм с эталонным генотипом CDC Frontier. Максимальная вариация (252 041 SNP) была обнаружена между Hat Trick и Slasher, тогда как минимальная вариация (145 415 SNP) наблюдалась между Hat Trick и Genesis90.В случае набора данных RAD максимальное количество SNP с эталонной последовательностью наблюдалось в ICC 4930. Наблюдалось, что общее количество SNP находится в диапазоне от 442 до 1151 между любой парой генотипов ().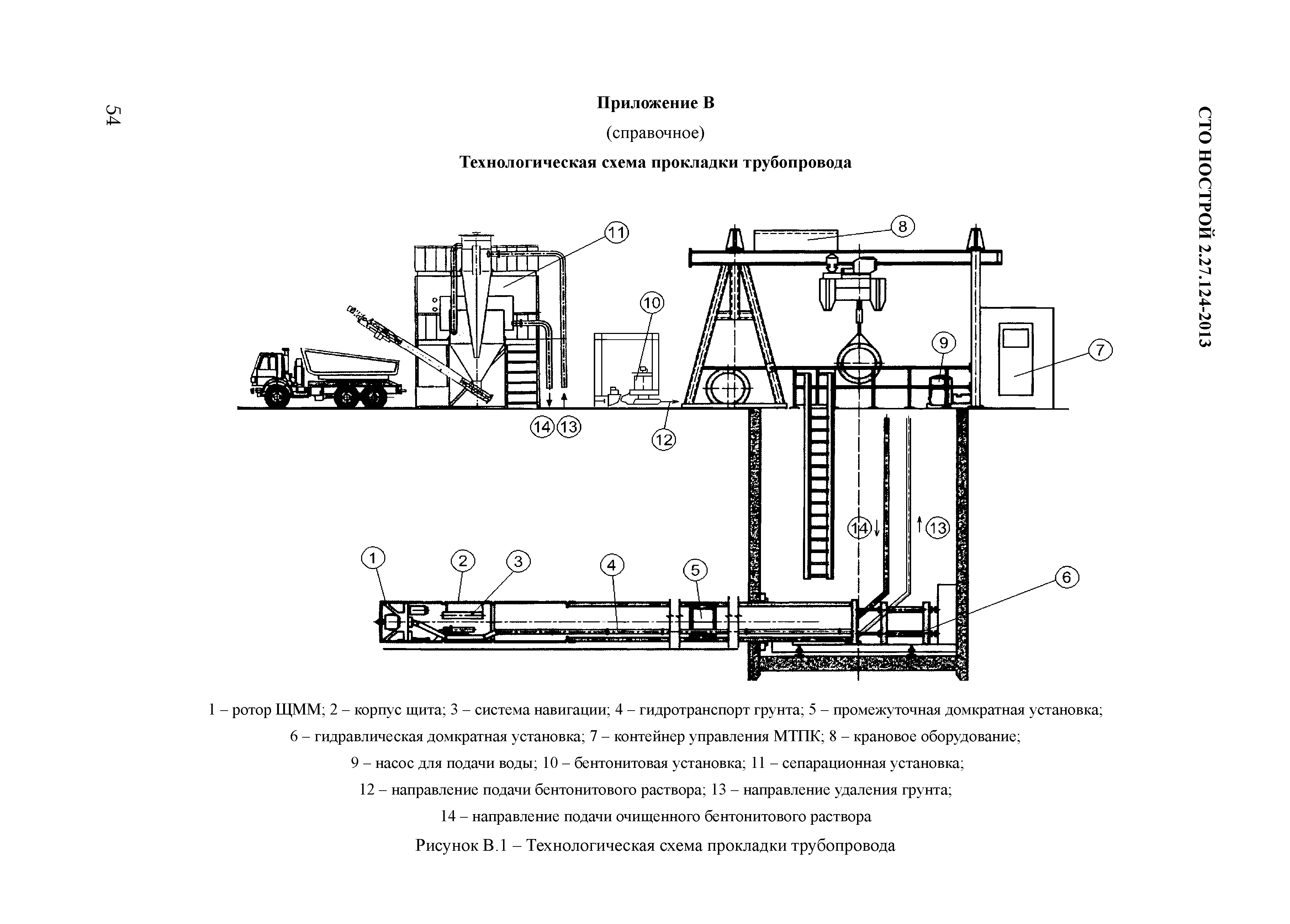 Была получена матрица SNP, состоящая из 28 348 полиморфных позиций по всем генотипам, включая эталон. Было обнаружено, что аллели в нескольких генотипах не типизированы, и, следовательно, они считались отсутствующими из-за отсутствия охвата или меньшего охвата. Это обычная характеристика данных секвенирования RAD, поэтому для преодоления этого ограничения можно выбрать вменение [54].В случае набора данных RNAseq HuaU606 показывает больше SNP по отношению к эталону, чем HuaU12 (). Однако между этими двумя генотипами было названо в общей сложности 13 294 SNP.
Была получена матрица SNP, состоящая из 28 348 полиморфных позиций по всем генотипам, включая эталон. Было обнаружено, что аллели в нескольких генотипах не типизированы, и, следовательно, они считались отсутствующими из-за отсутствия охвата или меньшего охвата. Это обычная характеристика данных секвенирования RAD, поэтому для преодоления этого ограничения можно выбрать вменение [54].В случае набора данных RNAseq HuaU606 показывает больше SNP по отношению к эталону, чем HuaU12 (). Однако между этими двумя генотипами было названо в общей сложности 13 294 SNP.
На диаграмме Венна показано распределение SNP, обнаруженных между четырьмя генотипами (Pistol, Hat Trick, Slasher и Genesis 90). В качестве эталонной последовательности использовали генотип CDC Frontier. Например, было обнаружено, что в общей сложности 95 329 SNP совпадают между генотипами Pistol и Hat Trick.Точно так же среди всех четырех генотипов было обнаружено, что 62 291 SNP являются общими.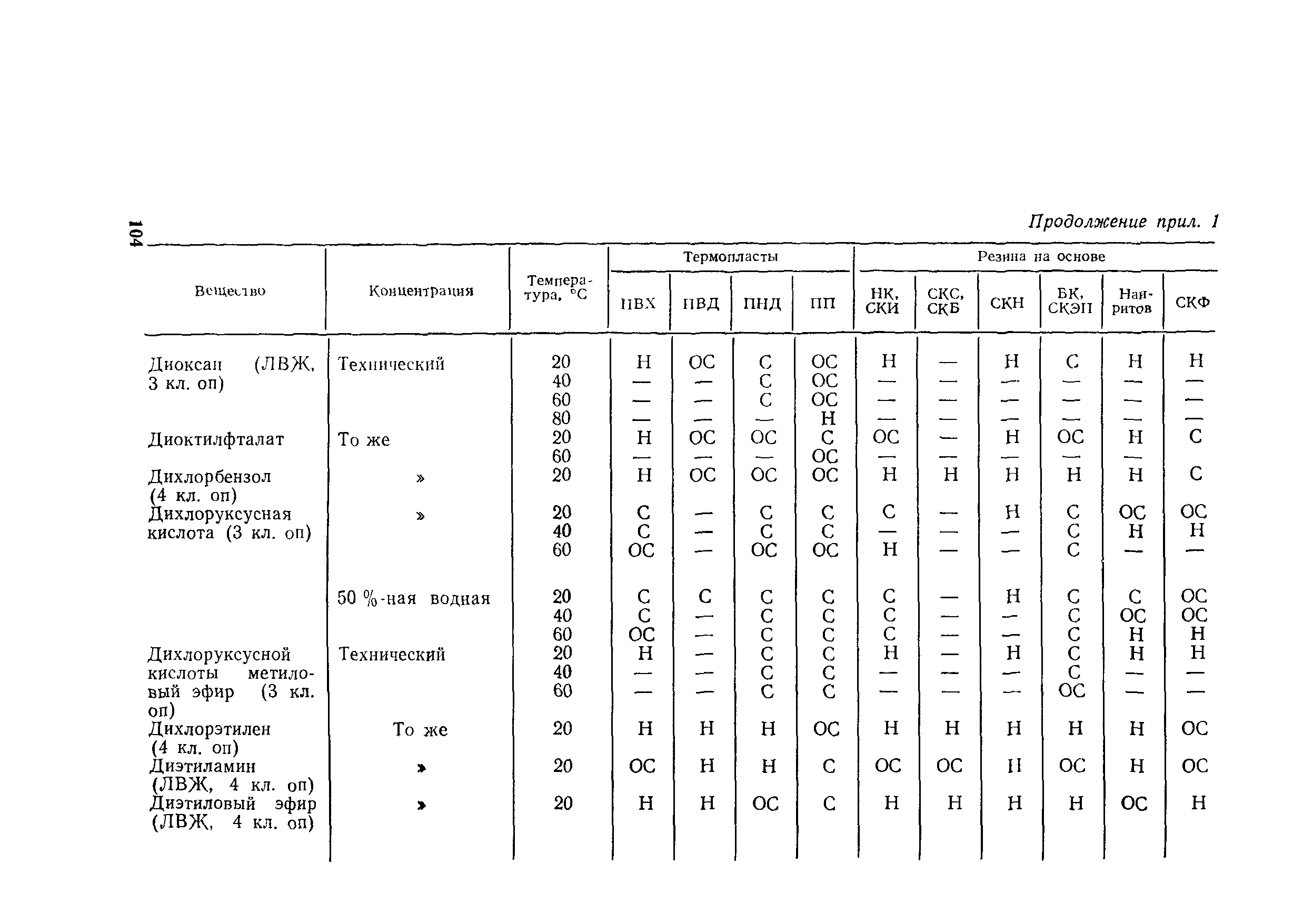
Таблица 4
Парное распределение SNP между генотипами, определенными в наборе данных RAD.
90 8367777| ICCV 03107 | МТП 4918 | МТП 4930 | МТП 4958 | МТП 5270 | ICCV 05530 | МТП 5810 | МТП 5912 | МТП 6263 | МТП 8261 | |||||
| Артикул | 5068 | 6250 | 9206 | 8502 | 7418 | 7461 | 8347 | 4985 | 6372 | 6372 | 5664 | |||
| ICCV 03107 | 442 | 470 | 599 | 501 | 499 | 528 | 528 | 455 | 667 | 471 | ||||
| МТП 4918 | 700 | 700 | 648 | 624 | 723 | 606 | 704 | 704 | 763 | 623 | ||||
| МТП 4930 | 1151 | 998 | 828 | 993 | 993 | 977 | 977 | 828 | ||||||
| МТП 4958 | 829 | 1016 | 852 | 892 | 752 | 752 | 617 | |||||||
| МТП 5270 | 945 | 886 | 791 | 783 | 613 | |||||||||
| ICCV 05530 | 972 | 778 | 924 | 774 | 774 | |||||||||
| МТП 5810 | 793 | 958 | 761 | 761 | ||||||||||
| МТП 5912 | 910 | 743 | ||||||||||||
| МТП 6263 | 581 |
Профилирование ASMU
ALLELE CONITING — это самая большая память интенсивной части трубопровода.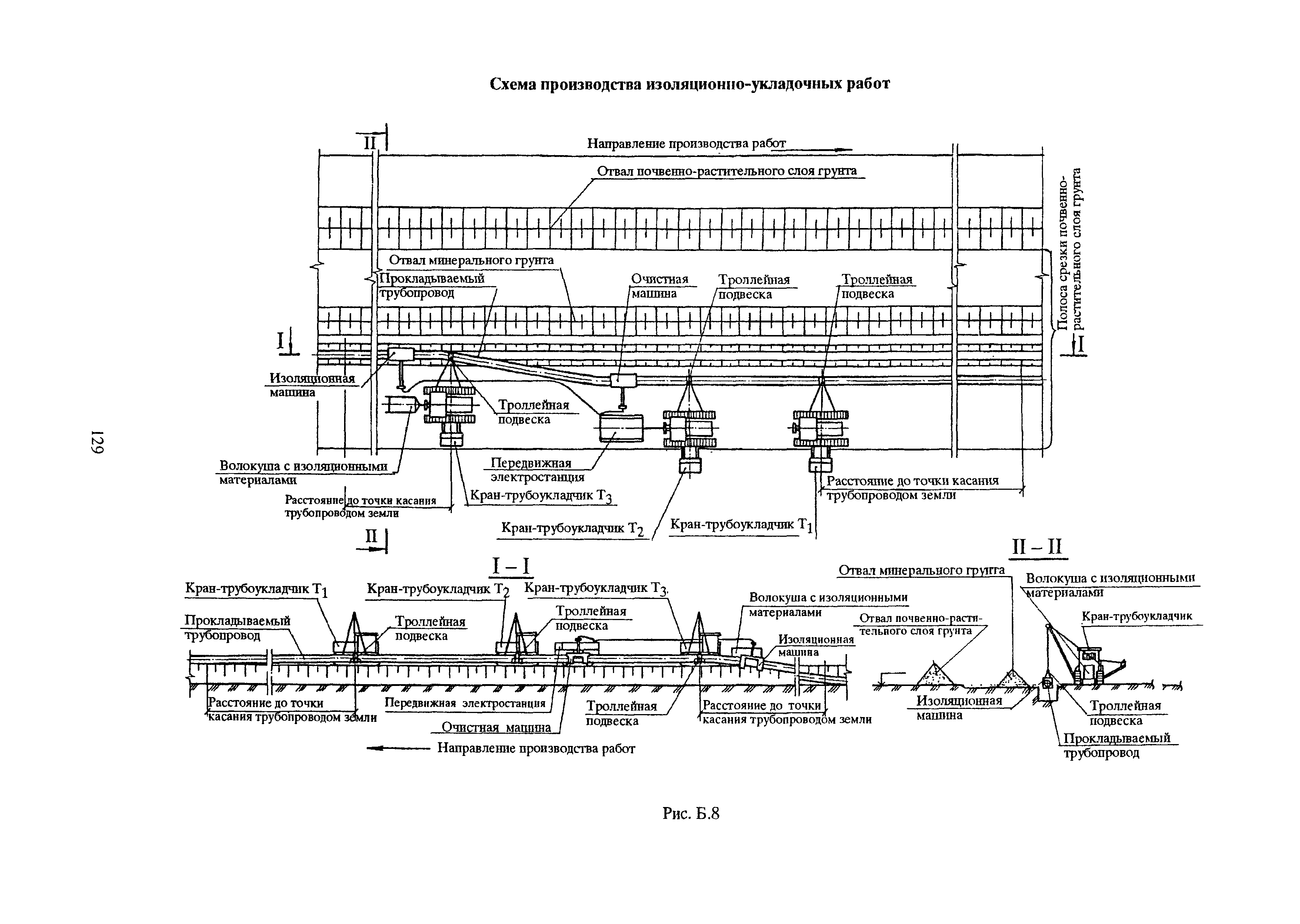 Был записан вычислительный профиль конвейера с тремя наборами данных, чтобы наблюдать пиковое потребление памяти, выделенное дисковое пространство и время, необходимое для завершения анализа. В каждом случае он был протестирован с 18 процессорами на настольном компьютере под управлением Linux с 48 ГБ ОЗУ (). Обнаружено, что набор данных RNAseq анализируется быстрее, чем RAD с последующим набором данных WGRS. Эталонные последовательности набора данных RNAseq сравнительно намного меньше, чем эталонные последовательности, используемые с наборами данных WGRS и RAD.Очевидно, что чем больше входных данных, тем больше времени требуется и, следовательно, требуется больше места на диске. Было обнаружено, что требуемая пиковая память выше для наборов данных WGRS и RAD по сравнению с набором данных RNAseq. Это связано с тем, что длины эталонных последовательностей в случае набора данных RNAseq намного меньше, чем в наборах данных WGRS и RAD. Кроме того, наборы данных WGRS и RAD в 17 и 3 раза больше, чем наборы данных RNAseq соответственно, и, следовательно, требования к памяти соответственно увеличиваются.
Был записан вычислительный профиль конвейера с тремя наборами данных, чтобы наблюдать пиковое потребление памяти, выделенное дисковое пространство и время, необходимое для завершения анализа. В каждом случае он был протестирован с 18 процессорами на настольном компьютере под управлением Linux с 48 ГБ ОЗУ (). Обнаружено, что набор данных RNAseq анализируется быстрее, чем RAD с последующим набором данных WGRS. Эталонные последовательности набора данных RNAseq сравнительно намного меньше, чем эталонные последовательности, используемые с наборами данных WGRS и RAD.Очевидно, что чем больше входных данных, тем больше времени требуется и, следовательно, требуется больше места на диске. Было обнаружено, что требуемая пиковая память выше для наборов данных WGRS и RAD по сравнению с набором данных RNAseq. Это связано с тем, что длины эталонных последовательностей в случае набора данных RNAseq намного меньше, чем в наборах данных WGRS и RAD. Кроме того, наборы данных WGRS и RAD в 17 и 3 раза больше, чем наборы данных RNAseq соответственно, и, следовательно, требования к памяти соответственно увеличиваются. Интересно, что набор данных RAD потребляет ресурсы памяти, аналогичные WGRS, несмотря на то, что он меньше по размеру, чем набор данных WGRS.Это связано с тем, что в набор данных RAD включено большее количество генотипов по сравнению с набором данных WGRS. Поэтому мы заключаем, что количество генотипов в качестве входных данных, размер эталонной хромосомы и количество данных влияют на объем оперативной памяти, требуемый конвейером. Следовательно, если у вас есть больше данных и/или нужно проанализировать больше генотипов, мы советуем соответственно запустить конвейер ISMU на ресурсоемких машинах. Однако данные транскриптома или геномы меньшего размера или меньшее количество генотипов больших геномов можно было бы эффективно обрабатывать на настольном компьютере.
Интересно, что набор данных RAD потребляет ресурсы памяти, аналогичные WGRS, несмотря на то, что он меньше по размеру, чем набор данных WGRS.Это связано с тем, что в набор данных RAD включено большее количество генотипов по сравнению с набором данных WGRS. Поэтому мы заключаем, что количество генотипов в качестве входных данных, размер эталонной хромосомы и количество данных влияют на объем оперативной памяти, требуемый конвейером. Следовательно, если у вас есть больше данных и/или нужно проанализировать больше генотипов, мы советуем соответственно запустить конвейер ISMU на ресурсоемких машинах. Однако данные транскриптома или геномы меньшего размера или меньшее количество генотипов больших геномов можно было бы эффективно обрабатывать на настольном компьютере.
Таблица 5
Профиль времени выполнения конвейера ISMU с тремя наборами данных (WGRS, RAD и RNAseq).
9| Datasets | WGRS | RAD | Секвенирование РНК | |||
| Метод (выравниватель-SNPcaller) | BWA-samtools | Bowtie-samtools | SOAP2-CbCC | |||
| Общее количество Ядра | 18 | 18 | 18 | 18 | 18 | |
| Общее количество генотипов | 4 | 10 | 2 | |||
| Размер входного файла (гигабайт) | 105 | 19.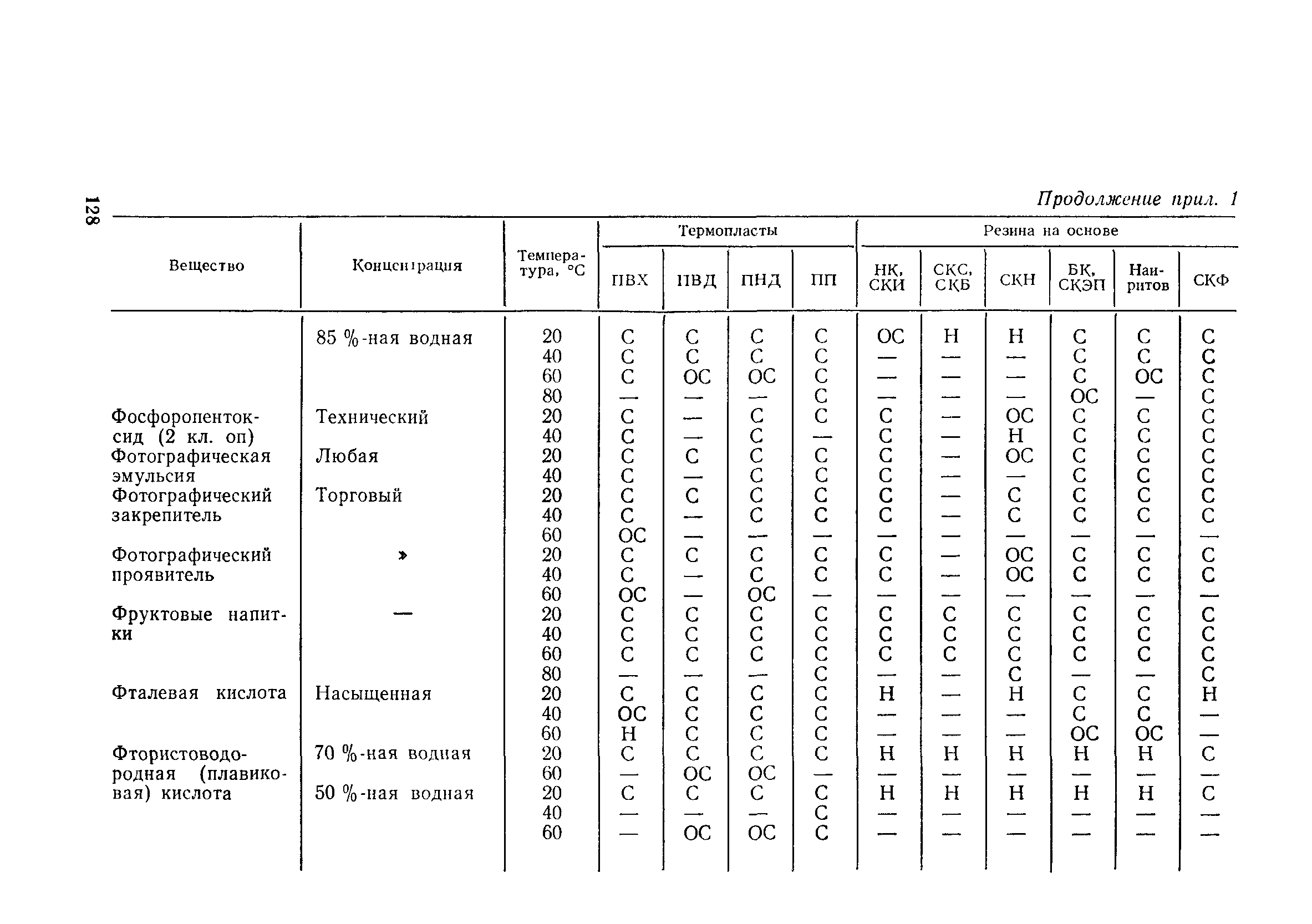 4 4 | 6.2 | 6.2 | ||
| Общее время (часы) | 26.25 | 4 9 | 2 | |||
| 250 | 57 | 17.5 | ||||
| Пиковая память (Gigabytes) | 45 | 48 | 3.6 |
Обсуждение
ISMU — это новый конвейер для добычи SNP с уникальными характеристиками, превосходящими другие конвейеры ().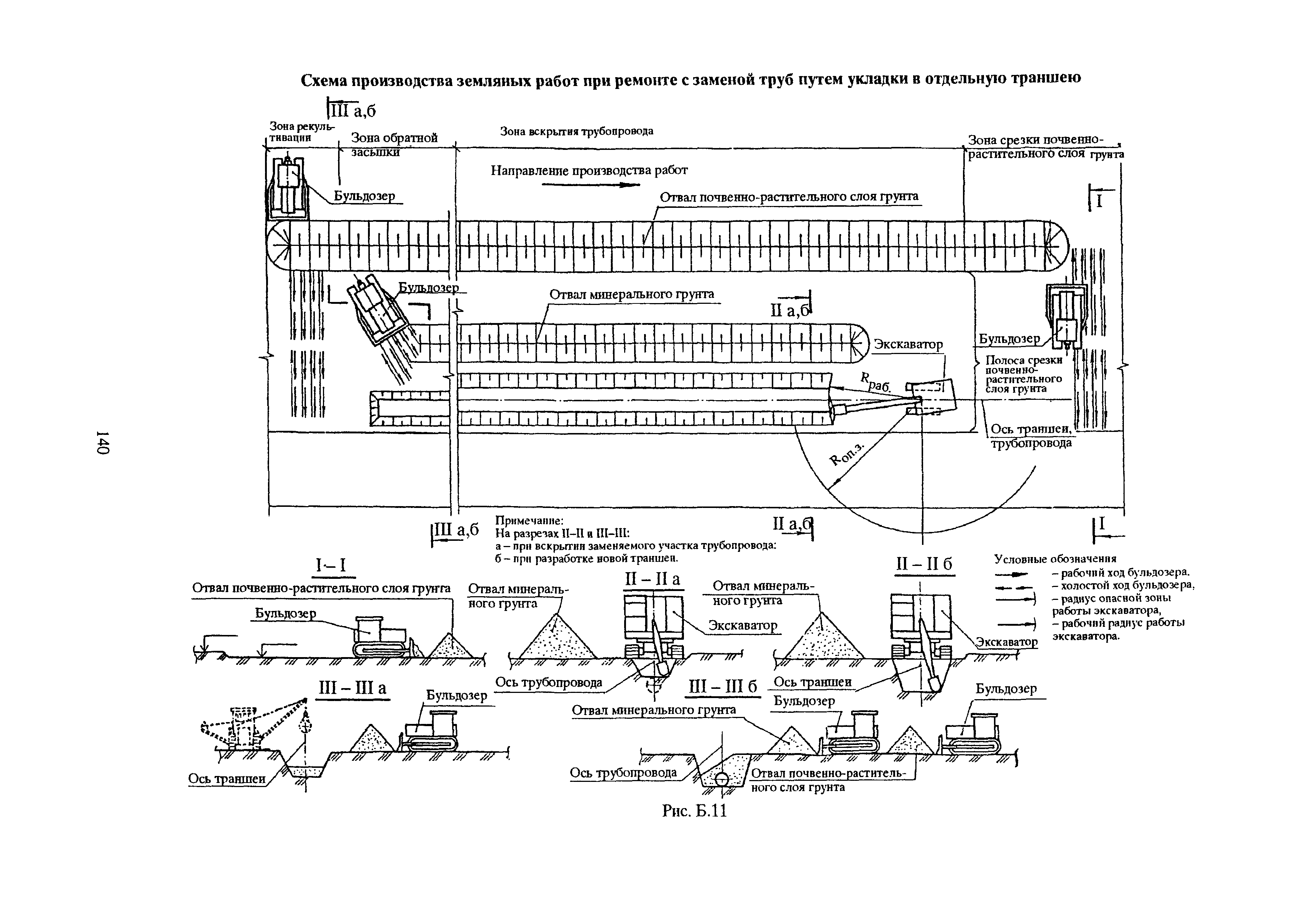 Как правило, существующие конвейеры для извлечения SNP основаны на командной строке, что требует технических знаний и, следовательно, не является удобным для пользователя.Следовательно, ISMU включает в себя графический пользовательский интерфейс (GUI), чтобы упростить анализ для сообщества молекулярных заводчиков, поскольку они не знакомы с техническими нюансами операционной системы Linux и инструментов командной строки. Интеграция надежных функций предварительной обработки и визуализации, особенно блинчиков для просмотра гаплотипов, делает его уникальным в своем роде конвейером. Кроме того, существующие конвейеры идентифицируют и сообщают SNP между эталоном и образцом, однако ISMU также напрямую сообщает об SNP между двумя образцами и предоставляет неизбыточный список SNP среди образцов.Это может быть использовано для сравнения и противопоставления аллельных различий между генотипами. Метод CbCC для обнаружения SNP является безэталонным методом, применимым для таких видов растений, для которых эталонный геном недоступен.
Как правило, существующие конвейеры для извлечения SNP основаны на командной строке, что требует технических знаний и, следовательно, не является удобным для пользователя.Следовательно, ISMU включает в себя графический пользовательский интерфейс (GUI), чтобы упростить анализ для сообщества молекулярных заводчиков, поскольку они не знакомы с техническими нюансами операционной системы Linux и инструментов командной строки. Интеграция надежных функций предварительной обработки и визуализации, особенно блинчиков для просмотра гаплотипов, делает его уникальным в своем роде конвейером. Кроме того, существующие конвейеры идентифицируют и сообщают SNP между эталоном и образцом, однако ISMU также напрямую сообщает об SNP между двумя образцами и предоставляет неизбыточный список SNP среди образцов.Это может быть использовано для сравнения и противопоставления аллельных различий между генотипами. Метод CbCC для обнаружения SNP является безэталонным методом, применимым для таких видов растений, для которых эталонный геном недоступен. Обнаруженные SNP также были представлены в виде электронной таблицы, содержащей гетерозиготность, значение PIC и распределение аллелей каждого маркера в разных генотипах. Это помогло бы селекционному сообществу расставить приоритеты SNP для генотипирования. SNP с высокими значениями PIC, как правило, демонстрируют высокий уровень полиморфизма в данном наборе зародышевой плазмы, что желательно для картирования сцепления, картирования QTL и исследований разнообразия, поэтому находят применение в молекулярной селекции.Фактически конвейер выводит частоту всех аллелей, присутствующих в образцах, и предоставляет селекционерам возможность использовать эту информацию для поиска новых и редких аллельных вариантов, которые могут иметь несколько функциональных значений для картирования ассоциаций признаков у сельскохозяйственных растений. Частота минорных аллелей (MAF) для локуса SNP может быть выведена непосредственно из частот аллелей. С другой стороны, более низкое значение гетерозиготности в локусе SNP дает ключ к обнаружению локуса с минорным аллелем в образцах.
Обнаруженные SNP также были представлены в виде электронной таблицы, содержащей гетерозиготность, значение PIC и распределение аллелей каждого маркера в разных генотипах. Это помогло бы селекционному сообществу расставить приоритеты SNP для генотипирования. SNP с высокими значениями PIC, как правило, демонстрируют высокий уровень полиморфизма в данном наборе зародышевой плазмы, что желательно для картирования сцепления, картирования QTL и исследований разнообразия, поэтому находят применение в молекулярной селекции.Фактически конвейер выводит частоту всех аллелей, присутствующих в образцах, и предоставляет селекционерам возможность использовать эту информацию для поиска новых и редких аллельных вариантов, которые могут иметь несколько функциональных значений для картирования ассоциаций признаков у сельскохозяйственных растений. Частота минорных аллелей (MAF) для локуса SNP может быть выведена непосредственно из частот аллелей. С другой стороны, более низкое значение гетерозиготности в локусе SNP дает ключ к обнаружению локуса с минорным аллелем в образцах. Кроме того, данные SNP, удовлетворяющие критериям платформы генотипирования KASP, фильтруются и предоставляются в формате ввода KASP для преобразования SNP в тесты KASP.Точно так же SNP, удовлетворяющие критериям платформы генотипирования Illumina, также были предоставлены в предварительном формате ввода. Это может быть непосредственно отправлено в онлайн-инструмент Illumina Assay Designing Tool (ADT) для оценки прогнозирования информации об успехе и статуса проверки [55]. Кроме того, если маркеры соответствуют пороговому баллу ADT, их можно использовать для разработки OPA (Oligo Pooled Assay) на чипах Bead для различных машин Illumina (BeadXpress, Infinium Assay).
Кроме того, данные SNP, удовлетворяющие критериям платформы генотипирования KASP, фильтруются и предоставляются в формате ввода KASP для преобразования SNP в тесты KASP.Точно так же SNP, удовлетворяющие критериям платформы генотипирования Illumina, также были предоставлены в предварительном формате ввода. Это может быть непосредственно отправлено в онлайн-инструмент Illumina Assay Designing Tool (ADT) для оценки прогнозирования информации об успехе и статуса проверки [55]. Кроме того, если маркеры соответствуют пороговому баллу ADT, их можно использовать для разработки OPA (Oligo Pooled Assay) на чипах Bead для различных машин Illumina (BeadXpress, Infinium Assay).
Таблица 6
Сравнение основных характеристик трубопровода ISMU с аналогичными трубопроводами.
908 73 Автономный| Особенности ИГМУ | SIMPLEX ngs- Магистральная | GATK INGAP | SeqGene ИГРЫ | TREAT Atlas2 | ||||||||
| Бесплатно | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | 9 |
| SE / PE обработка данных | Y / Y | Y / Y / N | Y / N | Д | Д/Д | н.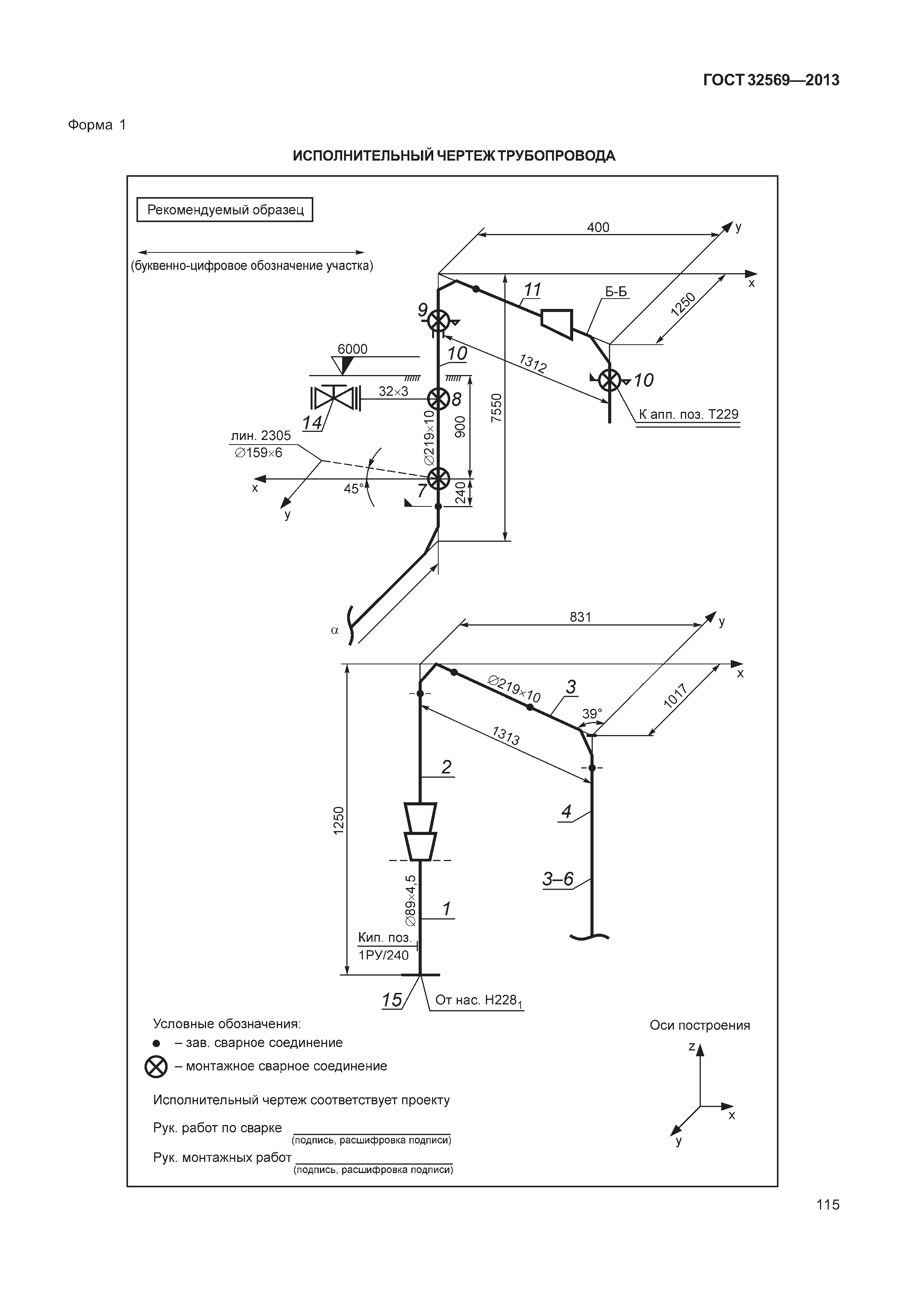 М М | Y / Y | Y / Y | Y / Y | Y / Y | ||
| NS / CS обработка данных | Y / N | Y / Y | Y / Y | Y / y | y / n | NM | Y / Y | Y / N | Y / N | Y / Y | Y / Y | |
| выравнивание | Y | Y | y | N | Y | Y | N | y | N | |||
| Количество центровочных инструментов | 5 | 1 | 1 | N | 2 | нет. м м | Н | 2 | Н | |||
| Вариант аннотаций | Да | Да | Н | Да | Н | Да | Да | Да | Н | |||
| Высоко настраиваемый | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | N | 3 N |
| гомо- / гетерозигозность | Y / Y | Y / y | N / N | Y / Y | N / N | Y / Y | N / Y | Y / Y | Y / Y | Y / Y | Y / Y | |
| y | y | y | y | y | n | Да | Н | Да | Н | |||
| графический пользовательский интерфейс | Да | Н | N | N | Да | Н | Н | Н | Да | |||
| Да | Да | Да | Да | Да | Да | Да | Да | Да | ||||
| поддержка ГПЦ | Да | Да | Да | Да | Н | Н | Н | Да | Н | |||
| поддержки пользователей Мульти | Да | Да | Н | Н | Н | Н | Н | Н | Н | |||
| Облако поддержка | N | Y | N | N | N | N | N | N | Y | Y | y |
На самом деле этот трубопровод был изначально разработан для транскриптовых данных, а затем продлен для геномных данные.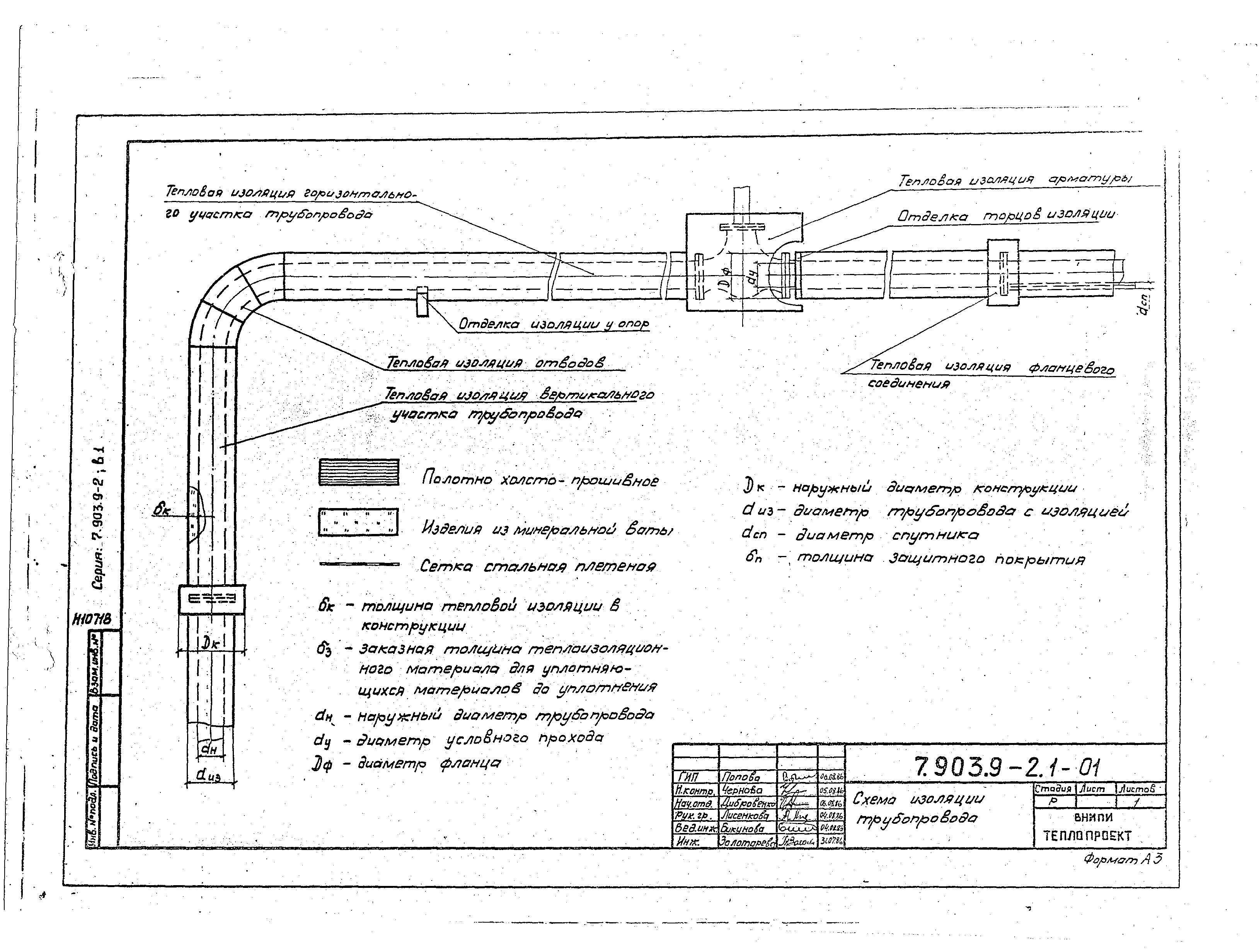 Конвейер был разработан для работы в средах с ограниченными ресурсами (небольшие лаборатории), что позволяет обрабатывать данные NGS даже при очень небольшом объеме оперативной памяти. Установка программного обеспечения и связанных с ним инструментов для обработки данных NGS и последующего анализа является сложной задачей для многих исследователей, особенно для молекулярного селекционера, который может иметь небольшой опыт в области вычислений. Поэтому ISMU также предоставляется в виде готового к использованию образа виртуальной машины, который позволяет пользователю быстро приступить к работе с настольным компьютером/рабочей станцией и с удобством добавить дополнительные аналитические возможности, если это необходимо.
Конвейер был разработан для работы в средах с ограниченными ресурсами (небольшие лаборатории), что позволяет обрабатывать данные NGS даже при очень небольшом объеме оперативной памяти. Установка программного обеспечения и связанных с ним инструментов для обработки данных NGS и последующего анализа является сложной задачей для многих исследователей, особенно для молекулярного селекционера, который может иметь небольшой опыт в области вычислений. Поэтому ISMU также предоставляется в виде готового к использованию образа виртуальной машины, который позволяет пользователю быстро приступить к работе с настольным компьютером/рабочей станцией и с удобством добавить дополнительные аналитические возможности, если это необходимо.
Выбор инструментов
Инструменты NGS с открытым исходным кодом были тщательно отобраны и интегрированы в ISMU. Конвейер предоставляет пользователю множество вариантов выравнивания и вызова SNP, что делает его универсальным и уникальным по сравнению с аналогичными конвейерами.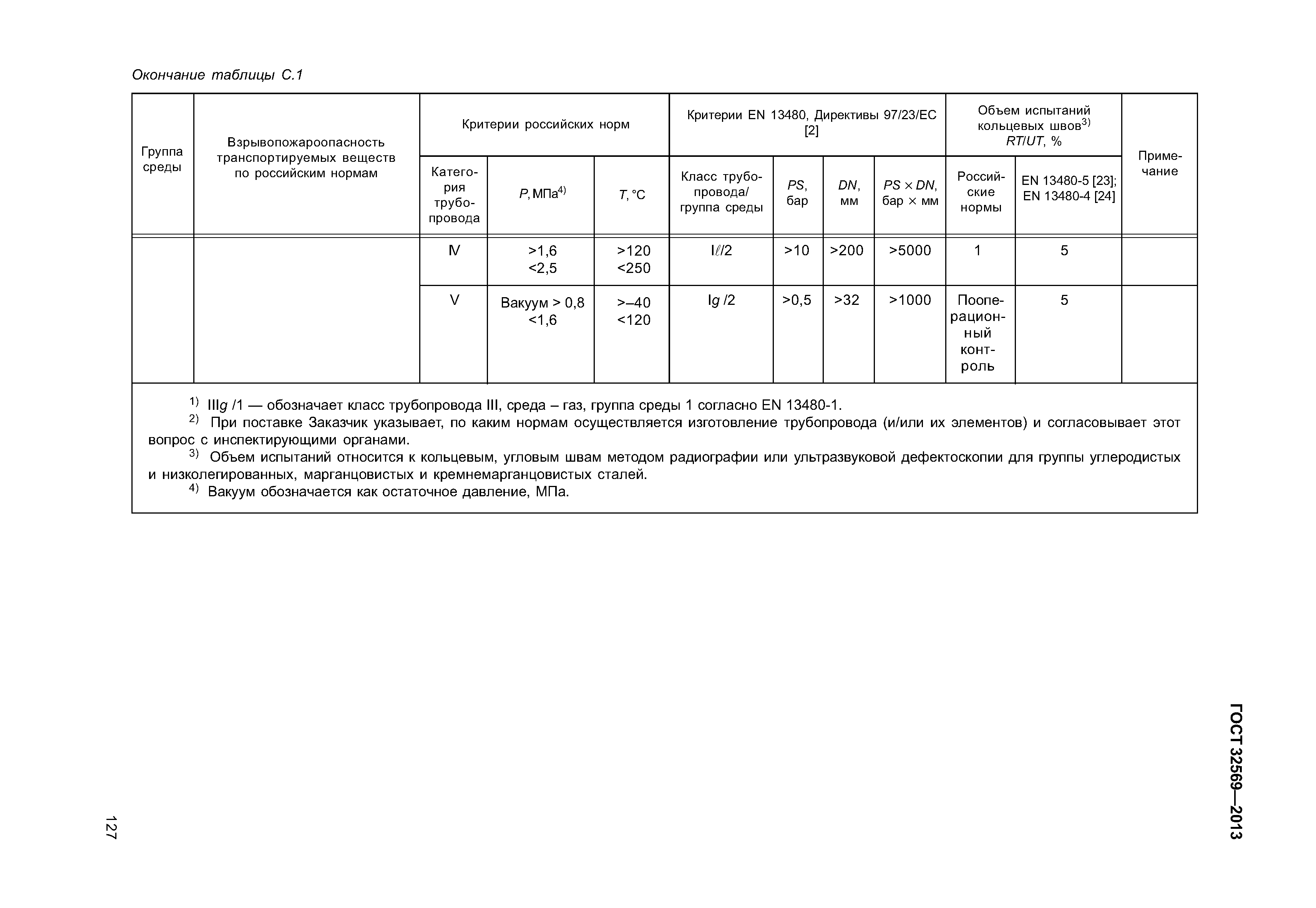 Несколько вариантов помогают пользователю провести повторный анализ с помощью различных инструментов, чтобы найти наилучшие или согласующиеся результаты. Однако у каждого инструмента есть свои плюсы и минусы. В случае, если доступны данные высокого качества, результаты с разными инструментами будут похожими, но с данными низкого качества (неглубокий охват) различия в результатах очевидны [48].
Несколько вариантов помогают пользователю провести повторный анализ с помощью различных инструментов, чтобы найти наилучшие или согласующиеся результаты. Однако у каждого инструмента есть свои плюсы и минусы. В случае, если доступны данные высокого качества, результаты с разными инструментами будут похожими, но с данными низкого качества (неглубокий охват) различия в результатах очевидны [48].
Выравнивание последовательности является одним из основных шагов, который сильно влияет на обнаружение маркера. На этом этапе инструменты сопоставления используют эвристику для согласования коротких чтений с эталонной последовательностью, поскольку исчерпывающие и точные алгоритмы не могут быть реализованы с вычислительной точки зрения. Первоначально небольшие области (начальные) в считываниях будут идентифицированы по сравнению с эталонной последовательностью, где с наибольшей вероятностью будет найдено место наилучшего совпадения. После определения меньшего подмножества возможных местоположений отображения на ограниченном подмножестве запускаются более медленные, но более точные алгоритмы выравнивания, такие как Смит-Уотерман [56]. Алгоритмы, которые используются для поиска небольшого набора потенциальных выравниваний в эталонной последовательности, можно разделить на две основные категории: хеш-таблица и суффиксное дерево/массив. Алгоритмы на основе хеширования требуют больше ресурсов (ОЗУ) и вычислительного времени (ELAND/Maq/SOAP). Массивы суффиксов в сочетании с методами сжатия, такими как преобразование Берроу-Уилера (BWT) и индекс FM, использовались для создания программ выравнивания, эффективных с точки зрения пространства и времени (BWA/BOWTIE/SOAP2), превосходящих методы на основе хэшей [56], [57].Всего в конвейер было интегрировано одно программное обеспечение на основе хеширования, Maq, и четыре программного обеспечения на основе BWT, Bowtie2, BWA, NovoAlign и SOAP2. Все инструменты способны обрабатывать короткие чтения, а также данные SE и PE; некоторое программное обеспечение, такое как BWA, также способно к длительному чтению. Следовательно, рекомендуется использовать варианты программного обеспечения на основе BWT в конвейере из-за быстрого времени работы и более низких требований к памяти.
Алгоритмы, которые используются для поиска небольшого набора потенциальных выравниваний в эталонной последовательности, можно разделить на две основные категории: хеш-таблица и суффиксное дерево/массив. Алгоритмы на основе хеширования требуют больше ресурсов (ОЗУ) и вычислительного времени (ELAND/Maq/SOAP). Массивы суффиксов в сочетании с методами сжатия, такими как преобразование Берроу-Уилера (BWT) и индекс FM, использовались для создания программ выравнивания, эффективных с точки зрения пространства и времени (BWA/BOWTIE/SOAP2), превосходящих методы на основе хэшей [56], [57].Всего в конвейер было интегрировано одно программное обеспечение на основе хеширования, Maq, и четыре программного обеспечения на основе BWT, Bowtie2, BWA, NovoAlign и SOAP2. Все инструменты способны обрабатывать короткие чтения, а также данные SE и PE; некоторое программное обеспечение, такое как BWA, также способно к длительному чтению. Следовательно, рекомендуется использовать варианты программного обеспечения на основе BWT в конвейере из-за быстрого времени работы и более низких требований к памяти.
Конвейер ISMU объединяет четыре программы вызова SNP. Программа SAMtools (v0.1.19) [47] была выбрана для вызова вариантов, поскольку это наиболее популярная программа вызова SNP, как следует из литературы.SAMtools принимает входные данные для центровки в формате SAM, созданные тремя программами для центровки, а именно BWA, Bowtie2 и NovoAlign. Однако выравнивания с использованием Maq и SOAP2 не сообщаются непосредственно в формате SAM, а скорее в формате Map, но могут быть преобразованы в формат SAM. Файлы формата карты при преобразовании в формат SAM не содержат информации заголовка. Этот заголовок может быть регенерирован из ссылки и предоставлен SAMtools для создания файлов BAM, которые можно использовать для вызова SNP. Однако такой файл BAM не работает должным образом с SAMtools.Следовательно, в качестве обходного пути были интегрированы программное обеспечение Maqsnp (скрипт в пакете Maq) и SOAPsnp [50] для вызова консенсуса и прогнозирования SNP на основе выравнивания SOAP и Maq соответственно.
В качестве альтернативы включен другой метод вызова SNP, консенсусный вызов на основе покрытия (CbCC) [48], который можно выбрать для использования с любым программным обеспечением для выравнивания, интегрированным в ISMU. Он использует файлы выравнивания в формате SAM, созданные с помощью инструментов выравнивания или преобразованные в формат SAM из других форматов.С другой стороны, CbCC был написан на Perl. Этот метод создает список SNP путем прямого сравнения двух генотипов и очень полезен при работе с видами, у которых отсутствует законченная эталонная последовательность генома.
Сравнение с существующим конвейером/программным обеспечением
Исследовательскому сообществу доступно несколько конвейеров с открытым исходным кодом для анализа данных NGS. Но не все из них поддерживают анализ необработанных данных NGS. Очень немногие конвейеры, такие как ISMU, начинают с предварительной обработки необработанных данных NGS, в то время как конвейеры, такие как GATK, GAMES, Atlas2 (), предназначены для работы, начиная с файлов выравнивания, таких как BAM/SAM. Многие конвейеры не включают этапы предварительной обработки, что влияет на прогнозируемую частоту ложных обнаружений SNP. Однако в ISMU этап предварительной обработки является необязательной, но рекомендуемой функцией, которую можно использовать в зависимости от качества входных данных. ISMU — это универсальный инструмент для анализа SNP, использующий необработанные данные в качестве входных данных с удобным интерфейсом (GUI), скрывающим вычислительные детали. Однако Atlas и inGAP — единственные другие конвейеры с графическим интерфейсом. На самом деле ISMU предназначен для помощи сообществу генетиков и селекционеров, которым работа с Linux/командной строкой кажется утомительной.Поэтому для предотвращения проблем с установкой на платформах Windows и Linux предоставляется виртуальный образ конвейера. ISMU предлагает широкий спектр инструментов выравнивания (Bowtie2, BWA, Maq, NovoAlign и SOAP2) и методов прогнозирования SNP (SAMtools, CbCC, SOAPsnp, Maq), которые редко доступны в других конвейерах и, следовательно, сами по себе являются уникальными функциями.
Многие конвейеры не включают этапы предварительной обработки, что влияет на прогнозируемую частоту ложных обнаружений SNP. Однако в ISMU этап предварительной обработки является необязательной, но рекомендуемой функцией, которую можно использовать в зависимости от качества входных данных. ISMU — это универсальный инструмент для анализа SNP, использующий необработанные данные в качестве входных данных с удобным интерфейсом (GUI), скрывающим вычислительные детали. Однако Atlas и inGAP — единственные другие конвейеры с графическим интерфейсом. На самом деле ISMU предназначен для помощи сообществу генетиков и селекционеров, которым работа с Linux/командной строкой кажется утомительной.Поэтому для предотвращения проблем с установкой на платформах Windows и Linux предоставляется виртуальный образ конвейера. ISMU предлагает широкий спектр инструментов выравнивания (Bowtie2, BWA, Maq, NovoAlign и SOAP2) и методов прогнозирования SNP (SAMtools, CbCC, SOAPsnp, Maq), которые редко доступны в других конвейерах и, следовательно, сами по себе являются уникальными функциями. Как правило, инструменты вызова SNP предоставляют SNP с оценкой качества/вероятности, тогда как ISMU обеспечивает доверительный коэффициент, отражающий глубину чтения.Этот коэффициент достоверности был бы полезен при маркировке SNP высокого качества для исследований генотипирования. Фактически ISMU является единственным конвейером, который имеет интегрированное средство для создания входного файла для разработки тестов для генотипирования. Конвейер предоставляет неизбыточный набор потенциальных SNP, который можно использовать в качестве входного файла для анализа KASP, а также для разработки OPA на основе Illumina для генотипирования. В то время как другие пайплайны предоставляют SNP только относительно эталона, ISMU дополнительно предоставляет SNP в попарных комбинациях генотипов, а также матрицу SNP, которая эквивалентна матрице SNP пайплайнов GBS (TASSEL) [58].ISMU может обрабатывать данные GBS/RAD и создавать результирующую матрицу со значением PIC и гетерозиготностью, что является еще одной уникальной функцией, отсутствующей в других конвейерах.
Как правило, инструменты вызова SNP предоставляют SNP с оценкой качества/вероятности, тогда как ISMU обеспечивает доверительный коэффициент, отражающий глубину чтения.Этот коэффициент достоверности был бы полезен при маркировке SNP высокого качества для исследований генотипирования. Фактически ISMU является единственным конвейером, который имеет интегрированное средство для создания входного файла для разработки тестов для генотипирования. Конвейер предоставляет неизбыточный набор потенциальных SNP, который можно использовать в качестве входного файла для анализа KASP, а также для разработки OPA на основе Illumina для генотипирования. В то время как другие пайплайны предоставляют SNP только относительно эталона, ISMU дополнительно предоставляет SNP в попарных комбинациях генотипов, а также матрицу SNP, которая эквивалентна матрице SNP пайплайнов GBS (TASSEL) [58].ISMU может обрабатывать данные GBS/RAD и создавать результирующую матрицу со значением PIC и гетерозиготностью, что является еще одной уникальной функцией, отсутствующей в других конвейерах.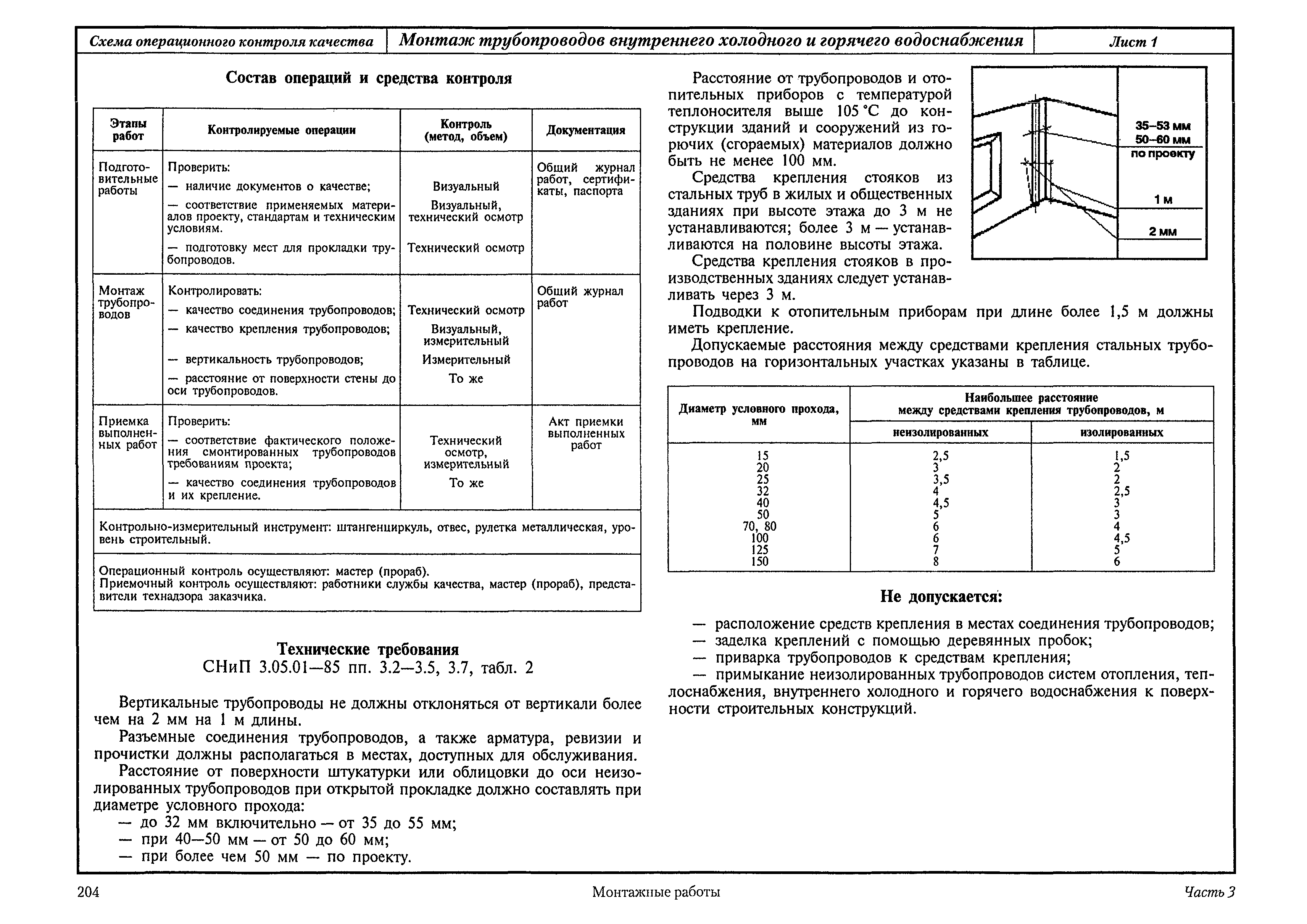 Как правило, конвейеры требуют от пользователя пошагового выполнения ряда команд для анализа, но ISMU делает этот процесс высокоавтоматизированным, так что пользователю нужно загрузить только входные данные, и результаты будут представлены.
Как правило, конвейеры требуют от пользователя пошагового выполнения ряда команд для анализа, но ISMU делает этот процесс высокоавтоматизированным, так что пользователю нужно загрузить только входные данные, и результаты будут представлены.
Конвейер предназначен для обработки наборов геномных (WGRS или RAD) и транскриптомных данных, которые включают как SE (один конец), так и PE (парный конец) чтения.Для облегчения последующего анализа представлена матрица SNP или матрица названных аллелей для всех генотипов. SNP, о которых сообщалось в результате, должны были считаться высоко достоверными. В наборе данных WGRS наблюдалось очень большое количество SNP со средней плотностью SNP 1,1/кб, в то время как в наборе данных RAD плотность SNP составляла 0,05/кб из-за характера технологий снижения сложности, которые не охватывают полную вариацию. В наборе данных RAD объем отсутствующей информации о генотипе в вызываемой позиции огромен и требует большей глубины секвенирования или вменения данных.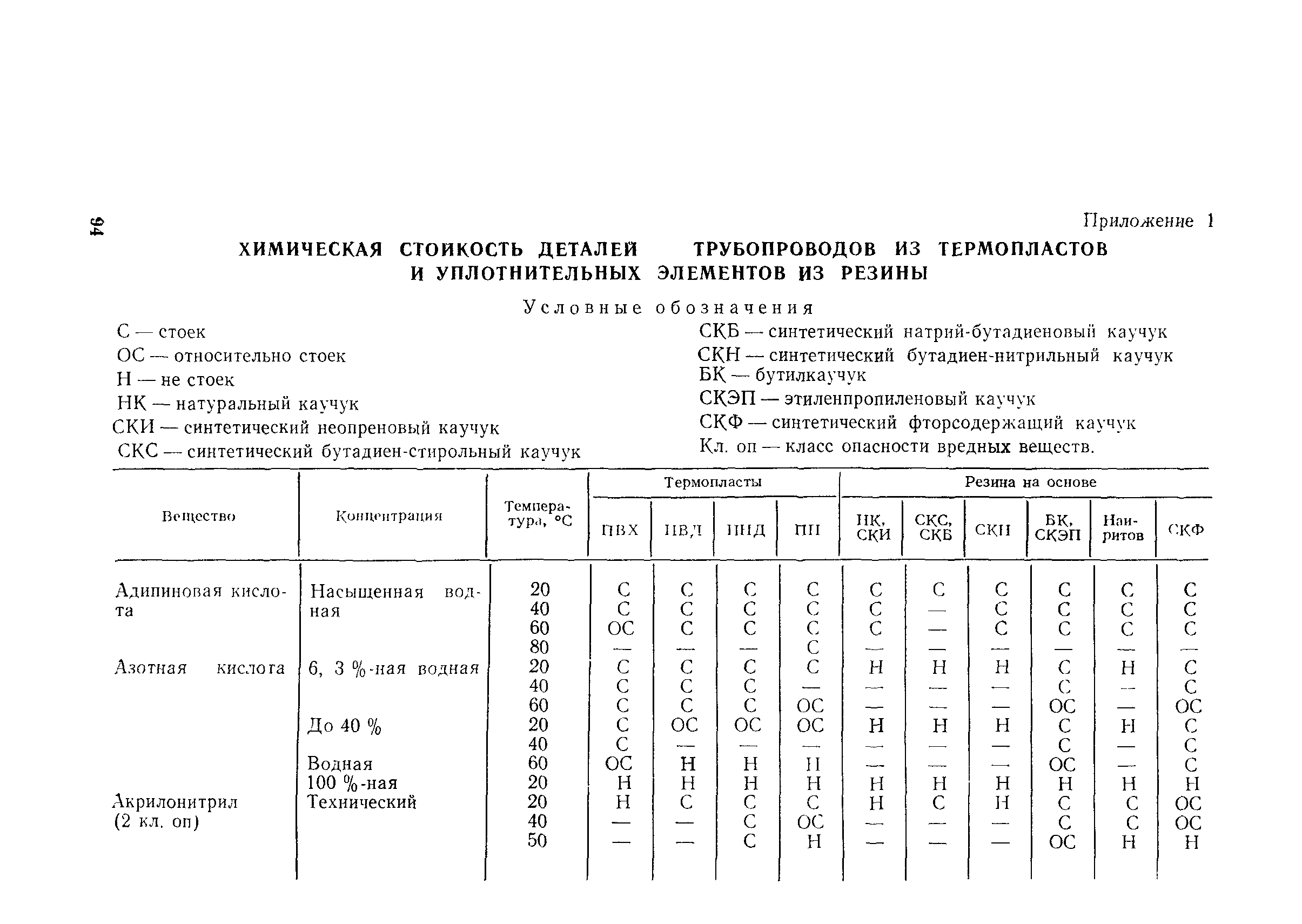 Для выравнивания набора данных RNAseq использовали SOAP2, который не допускает разрывов или отсечения на концах прочтений при выравнивании. Из-за этого строгого режима выравнивания SOAP2 выравнивал меньшую часть прочтений с удаленными эталонными последовательностями (унигенными), что проявляется в меньшем количестве обнаруженных SNP по сравнению с методами, основанными на выравнивании с пробелами [43].
Для выравнивания набора данных RNAseq использовали SOAP2, который не допускает разрывов или отсечения на концах прочтений при выравнивании. Из-за этого строгого режима выравнивания SOAP2 выравнивал меньшую часть прочтений с удаленными эталонными последовательностями (унигенными), что проявляется в меньшем количестве обнаруженных SNP по сравнению с методами, основанными на выравнивании с пробелами [43].
Заключение
ISMU — это проверенный конвейер добычи SNP для данных NGS из одной или нескольких выборок. Основные функции конвейера включают графический интерфейс, распараллеливание, несколько инструментов анализа и многопользовательскую поддержку для облегчения анализа огромных данных NGS для экспериментов по генотипированию.Хотя он был разработан для использования с сельскохозяйственными культурами (например, нутом, голубиным горохом и т. д.), его также можно использовать с данными о животных и микробах. Следовательно, данные могут обрабатываться независимо от организма. Простой в использовании графический интерфейс побуждает ученых/исследователей, не занимающихся биоинформатикой, анализировать данные в своих лабораториях на настольных компьютерах. ISMU объединяет проверенные инструменты анализа NGS и предлагает несколько вариантов выравнивания и вызова SNP. Он включает в себя полный рабочий процесс от предварительной обработки необработанных данных до расшифровки SNP и сообщения маркеров для экспериментов по генотипированию.Конвейер выводит информацию об вариациях проанализированных образцов в простом формате для последующих анализов, таких как исследования генотипирования. Распараллеливание ISMU позволяет эффективно использовать современные настольные компьютеры с несколькими ядрами. Полное приложение тщательно протестировано и также распространяется на компакт-дисках, а также в виде готового к использованию образа виртуальной коробки.
Простой в использовании графический интерфейс побуждает ученых/исследователей, не занимающихся биоинформатикой, анализировать данные в своих лабораториях на настольных компьютерах. ISMU объединяет проверенные инструменты анализа NGS и предлагает несколько вариантов выравнивания и вызова SNP. Он включает в себя полный рабочий процесс от предварительной обработки необработанных данных до расшифровки SNP и сообщения маркеров для экспериментов по генотипированию.Конвейер выводит информацию об вариациях проанализированных образцов в простом формате для последующих анализов, таких как исследования генотипирования. Распараллеливание ISMU позволяет эффективно использовать современные настольные компьютеры с несколькими ядрами. Полное приложение тщательно протестировано и также распространяется на компакт-дисках, а также в виде готового к использованию образа виртуальной коробки.
ISMU нацелен на сообщество специалистов по молекулярной селекции растений, не обладающих достаточными знаниями в области вычислений. Селекционеры/исследователи растений потенциально могли бы использовать его для быстрой обработки данных и, таким образом, получения биологического понимания генетических событий.SNP, обнаруженные конвейером, можно использовать в таких приложениях, как картирование связей, картирование признаков, TILLING, GWAS и QTLSeq (объемный сегрегантный анализ).
Селекционеры/исследователи растений потенциально могли бы использовать его для быстрой обработки данных и, таким образом, получения биологического понимания генетических событий.SNP, обнаруженные конвейером, можно использовать в таких приложениях, как картирование связей, картирование признаков, TILLING, GWAS и QTLSeq (объемный сегрегантный анализ).
Материалы и методы
Наборы данных о последовательности
Для оценки эффективности недавно разработанного конвейера ISMU были собраны два набора геномных данных по нуту и один набор транскриптомных данных растений по арахису. Первый набор данных состоит из данных полногеномного повторного секвенирования (WGRS) четырех генотипов нута, а именно Pistol, Slasher, Hat Trick и Genesis 90, как сообщается в Varshney et al.[59]. Вторым набором данных был набор данных секвенирования RAD из десяти генотипов нута, а именно ICCV 03107, ICC 4918, ICC 4930, ICC 4958, ICC 5270, ICCV 05530, ICC 5810, ICC 5912, ICC 6263 и ICC 8261 [59]. Эти наборы данных доступны по адресу http://hpc.icrisat.cgiar.org/ISMU/datasets. Третий набор данных включает данные транскриптома (RNAseq) двух генотипов арахиса HuaU12 (SRR647081) и HuaU606 (SRR647076), загруженные из базы данных архива считывания последовательностей (SRA) NCBI. Наборы данных WGRS и RNAseq представляли собой данные с парным концом (PE), тогда как набор данных RAD был с одним концом (SE).Наборы данных WGRS и RAD используют черновую последовательность генома нута [59] в качестве эталонной последовательности, в то время как набор данных RNAseq использует репрезентативные последовательности unigene арахиса, загруженные из базы данных UniGene (ftp://ftp.ncbi.nih.gov/repository/UniGene/Arachis_hypogea/ Ahy.seq.uniq.gz).
Эти наборы данных доступны по адресу http://hpc.icrisat.cgiar.org/ISMU/datasets. Третий набор данных включает данные транскриптома (RNAseq) двух генотипов арахиса HuaU12 (SRR647081) и HuaU606 (SRR647076), загруженные из базы данных архива считывания последовательностей (SRA) NCBI. Наборы данных WGRS и RNAseq представляли собой данные с парным концом (PE), тогда как набор данных RAD был с одним концом (SE).Наборы данных WGRS и RAD используют черновую последовательность генома нута [59] в качестве эталонной последовательности, в то время как набор данных RNAseq использует репрезентативные последовательности unigene арахиса, загруженные из базы данных UniGene (ftp://ftp.ncbi.nih.gov/repository/UniGene/Arachis_hypogea/ Ahy.seq.uniq.gz).
Разработка конвейера
Инструменты для выравнивания (BWA, Maq, Bowtie2, NovoAlign и SOAP2), вызова SNP (Samtools и CbCC), визуализации SNP (Tablet, Flapjack) и анализа распределения аллелей (Flapjack) были распакованы и скомпилирован на Redhat Linux.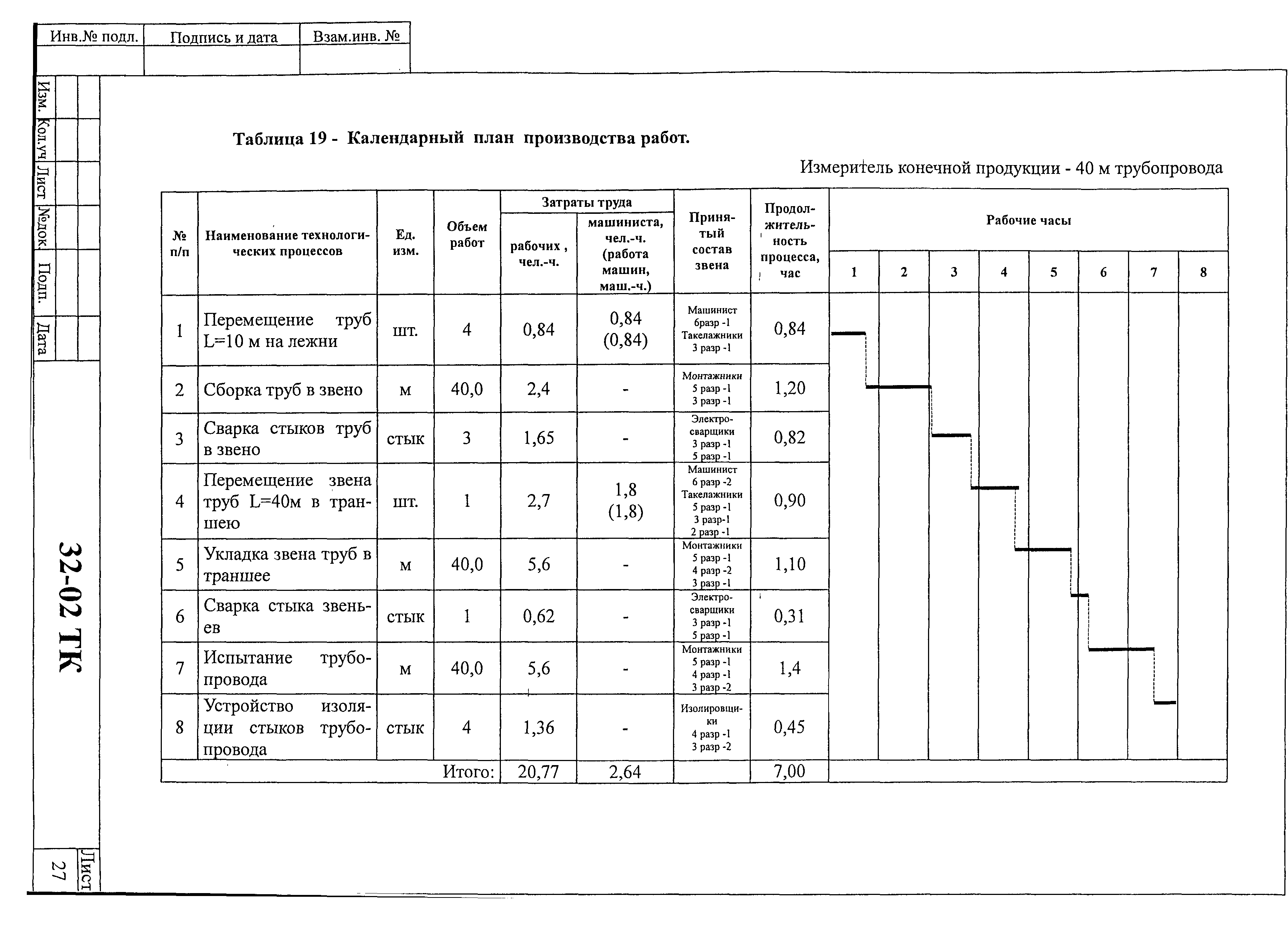 Затем эти инструменты были интегрированы в конвейер с использованием языка Perl. В этом процессе было написано несколько внутренних сценариев perl/bash, которые были встроены в конвейер, чтобы обеспечить автоматизацию, функциональность и визуализацию. Эти сценарии использовались для обеспечения необходимых функций, таких как предварительная обработка данных, создание входных файлов для Flapjack, обработка SNP для расчета частоты аллеля, значения PIC и создание файлов для разработки анализов Illumina и KASPar. Большинство сценариев были написаны с возможностью многопоточности для быстрого выполнения анализа при обработке больших наборов данных.Для разработки конвейера GUI был разработан интерактивный интерфейс, связанный с загрузкой данных, выбором инструментов, настройкой параметров анализа, расположением анализируемых данных и форматом вывода. Ввод, вводимый пользователем через графический интерфейс, передается в основной сценарий конвейера, который автоматически генерирует сценарий оболочки для выполнения различных шагов конвейера.
Затем эти инструменты были интегрированы в конвейер с использованием языка Perl. В этом процессе было написано несколько внутренних сценариев perl/bash, которые были встроены в конвейер, чтобы обеспечить автоматизацию, функциональность и визуализацию. Эти сценарии использовались для обеспечения необходимых функций, таких как предварительная обработка данных, создание входных файлов для Flapjack, обработка SNP для расчета частоты аллеля, значения PIC и создание файлов для разработки анализов Illumina и KASPar. Большинство сценариев были написаны с возможностью многопоточности для быстрого выполнения анализа при обработке больших наборов данных.Для разработки конвейера GUI был разработан интерактивный интерфейс, связанный с загрузкой данных, выбором инструментов, настройкой параметров анализа, расположением анализируемых данных и форматом вывода. Ввод, вводимый пользователем через графический интерфейс, передается в основной сценарий конвейера, который автоматически генерирует сценарий оболочки для выполнения различных шагов конвейера. Во время выполнения сценарий оболочки вызывает другое программное обеспечение и встроенные сценарии поэтапно, в то время как окно графического интерфейса показывает состояние выполнения.Кроме того, он также предоставляет возможность прекратить выполнение конвейера на любом этапе. В конце выполнения конвейера появляется другая страница, показывающая варианты загрузки и отображения различных выходных данных. Конвейер сохраняет все выходные файлы в предоставленной пользователем выходной папке, которую пользователь может получить на более позднем этапе.
Во время выполнения сценарий оболочки вызывает другое программное обеспечение и встроенные сценарии поэтапно, в то время как окно графического интерфейса показывает состояние выполнения.Кроме того, он также предоставляет возможность прекратить выполнение конвейера на любом этапе. В конце выполнения конвейера появляется другая страница, показывающая варианты загрузки и отображения различных выходных данных. Конвейер сохраняет все выходные файлы в предоставленной пользователем выходной папке, которую пользователь может получить на более позднем этапе.
Оценка пайплайна
Выбранные наборы данных были подвергнуты качественной фильтрации с целью отбраковки низкокачественных прочтений, т. е. прочтений, содержащих более 30% оснований низкого качества (Q<20).Эти отфильтрованные чтения были обрезаны с обоих концов, что привело к уменьшению длины чтения. Результирующие парные чтения концов не будут одинаковыми по длине. Чтения, прошедшие эти фильтры качества в наборах данных WGRS, RAD и RNAseq, затем были сопоставлены с эталоном с использованием различных инструментов выравнивания в ISMU. Наборы данных WGRS и наборы данных RAD были сопоставлены с геномом нута с использованием BWA и Bowtie2 соответственно. Набор данных RNAseq был сопоставлен с unigenes арахиса с использованием SOAP2. Samtools использовался для вызова вариантов для наборов данных WGRS и RAD, а CbCC использовался с набором данных RNAseq.Все наборы данных были проанализированы на 24-ядерной машине Linux с 48 ГБ ОЗУ.
Наборы данных WGRS и наборы данных RAD были сопоставлены с геномом нута с использованием BWA и Bowtie2 соответственно. Набор данных RNAseq был сопоставлен с unigenes арахиса с использованием SOAP2. Samtools использовался для вызова вариантов для наборов данных WGRS и RAD, а CbCC использовался с набором данных RNAseq.Все наборы данных были проанализированы на 24-ядерной машине Linux с 48 ГБ ОЗУ.
Заявление о финансировании
Программа CGIAR Generation Challenge (GCP), Мексика финансировала этот проект. Эта работа проводилась в рамках Исследовательской программы КГМСХИ по зерновым культурам в засушливых районах и Исследовательской программы КГМСХИ по зернобобовым культурам. Спонсоры не участвовали в разработке исследования, сборе и анализе данных, принятии решения о публикации или подготовке рукописи.
Ссылки
1. Имельфорт М., Эдвардс Д. (2009) De novo секвенирование геномов растений с использованием технологий второго поколения.Кратко Биоинформ 10: 609–618 10.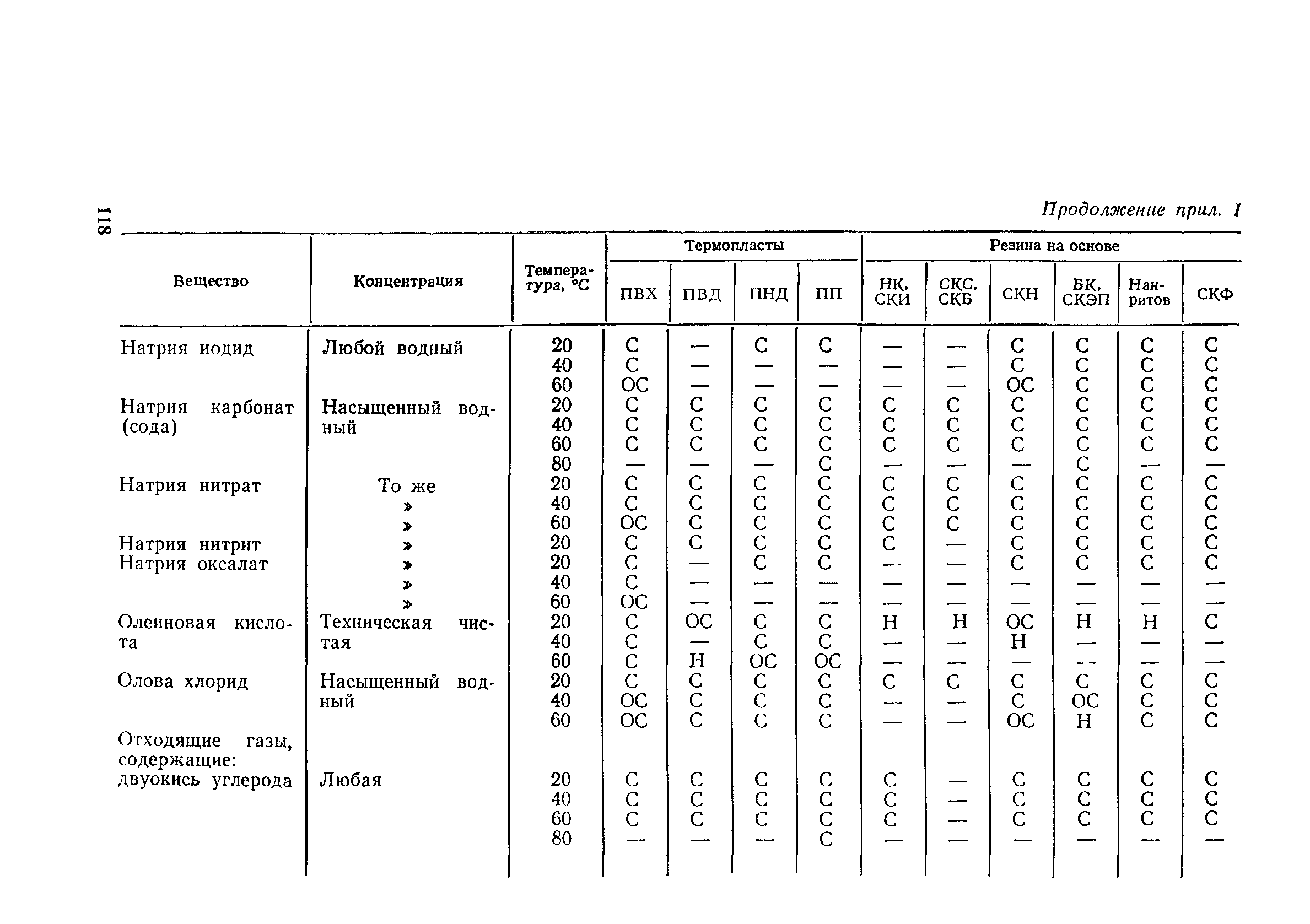 1093/нагрудник/bbp039
[PubMed] [CrossRef] [Google Scholar]2.
Варшней Р.К., Наяк С.Н., Мэй Г.Д., Джексон С.А. (2009)Технологии секвенирования следующего поколения и их значение для генетики и селекции сельскохозяйственных культур. Тенденции Биотехнологии
27: 522–530
10.1016/j.tibtech.2009.05.006
[PubMed] [CrossRef] [Google Scholar]3.
Маршалл Д.Дж., Хейворд А., Илз Д., Имельфорт М., Стиллер Дж. и др. (2010) Целевая идентификация геномных регионов с использованием TAGdb. Растительные методы
6: 19–19
10.1186/1746-4811-6-19
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]4.Майкл Т.П., Джексон С. (2013) Первые 50 геномов растений. Plant Gen 6: doi:10.3835/plantgenome2013.03.0001 [Google Scholar]5.
Дюран С., Эпплби Н., Варди М., Имельфорт М., Эдвардс Д. и др. (2009)Обнаружение полиморфизма одиночных нуклеотидов в ячмене с использованием autoSNPdb. Завод Биотехнолог J
7: 326–333
10.1111/j.1467-7652.2009.00407.x
[PubMed] [CrossRef] [Google Scholar]6.
Лу Т., Лу Г., Фан Д., Чжу С., Ли В. и др. (2010) Функциональная аннотация транскриптома риса при разрешении одного нуклеотида с помощью RNA-seq.Геном Res
20: 1238–1249 гг.
10.1101/гр.106120.110
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]7.
Дубей А., Фармер А., Шлютер Дж., Кэннон С.Б., Абернати Б. и др. (2011)Определение сборки транскриптома и ее использование для динамики генома и исследований профилирования транскриптома у голубиного гороха ( Cajanus cajan L.). ДНК Res
18: 153–164
10.1093/dnares/dsr007
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]8.
Гарг Р., Патель Р.К., Тьяги А.К., Джейн М. (2011) Сборка транскриптома нута De novo с использованием коротких прочтений для обнаружения генов и идентификации маркеров.ДНК Res
18: 53–63
10.1093/dnares/dsq028
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]9.
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, et al. (2011) Крупномасштабный транскриптомный анализ нута ( Cicer arietinum L.), бесхозной бобовой культуры полузасушливых тропиков Азии и Африки. Завод Биотехнолог J
9: 922–931
10.1111/j.1467-7652.2011.00625.x
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]10.
Дешам С., Ла Рота М., Раташак Дж. П., Биддл П., Турин Д. и др.(2010) Быстрое обнаружение однонуклеотидного полиморфизма по всему геному у сои и риса путем глубокого повторного секвенирования библиотек с уменьшенным представлением с помощью анализатора генома Illumina. Ген завода
3: 53–68
10.3835/геном растений2009.09.0026
[Перекрестная ссылка] [Академия Google] 11.
ван Тассел К.П., Смит Т.П.Л., Матукумалли Л.К., Тейлор Дж.Ф., Шнабель Р.Д. и соавт. (2008)Обнаружение SNP и оценка частоты аллелей путем глубокого секвенирования библиотек с уменьшенным представлением. Нат Методы
5: 247–252
10.1038/н-мет.1185
[PubMed] [CrossRef] [Google Scholar] 12.Гор М.А., Чиа Дж.М., Элшир Р.Дж., Сан К., Эрсоз Э.С. и др. (2009) Карта гаплотипов кукурузы первого поколения. Наука
326: 1115–1117
10.1126/наука.1177837
[PubMed] [CrossRef] [Google Scholar] 13.
Hyten DL, Cannon SB, Song Q, Weeks N, Fickus EW и др. (2010)Обнаружение SNP с высокой пропускной способностью посредством глубокого повторного секвенирования библиотеки с уменьшенным представлением для закрепления и ориентации каркасов в последовательности всего генома сои. Геномика BMC
11: 38–38
10.1186/1471-2164-11-38
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]15.van Orsouw NJ, Hogers RCJ, Janssen A, Yalcin F, Snoeijers S, et al. (2007) Снижение сложности полиморфных последовательностей (CRoPS): новый подход к обнаружению крупномасштабного полиморфизма в сложных геномах. PLoS один
2: e1172
10.1371/journal.pone.0001172
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]16.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, et al. (2008)Быстрое открытие SNP и генетическое картирование с использованием секвенированных маркеров RAD. PLoS один
3: e3376
10.1371/journal.pone.0003376
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]17.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, et al. (2011) Надежный подход простого генотипирования путем секвенирования (GBS) для видов с большим разнообразием. PLoS один
6: e19379
10.1371/journal.pone.0019379
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]18.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, et al. (2011)Полногеномное открытие генетических маркеров и генотипирование с использованием секвенирования следующего поколения. Нат Рев Жене
2: 499–510
10.1038/нрг3012
[PubMed] [CrossRef] [Google Scholar] 19.Huang X, Feng Q, Qian Q, Zhao Q, Wang L и др. (2009) Высокопроизводительное генотипирование путем полногеномного повторного секвенирования. Геном Res
19: 1068–1076
10.1101/гр.089516.108
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]20.
Мецкер М.Л. (2010) Технологии секвенирования — следующее поколение. Нат Рев Жене
11: 31–46
10.1038/nrg2626
[PubMed] [CrossRef] [Google Scholar] 22.
You FM, Huo N, Deal KR, Gu YQ, Luo MC и др. (2011)Обнаружение полногеномных SNP на основе аннотаций в большом и сложном геноме Aegilops tauschii с использованием секвенирования следующего поколения без эталонной последовательности генома.Геномика BMC
12: 59–59
10.1186/1471-2164-12-59
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]23.
Гаур Р., Азам С., Джина Дж., Хан А.В., Чоудхари С. и др. (2012)Обнаружение и генотипирование SNP с высокой пропускной способностью для построения карты насыщенного сцепления нута ( Cicer arietinum L.). ДНК Res
19: 357–373
10.1093/dnares/dss018
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]24.
Хаффорд М.Б., Сюй С., ван Хеерваарден Дж., Пихаярви Т., Чиа Дж.-М. и др. (2012) Сравнительная популяционная геномика одомашнивания и улучшения кукурузы.Нат Жене
44: 808–811
10.1038/нг.2309
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]25.
Xu X, Liu X, Ge S, Jensen JD, Hu F, et al. (2012) Повторное секвенирование 50 образцов культурного и дикого риса дает маркеры для идентификации агрономически важных генов. Нат Биотехнолог
30: 105–111
10.1038/нбт.2050
[PubMed] [CrossRef] [Google Scholar] 26.
Lam H-M, Xu X, Liu X, Chen W, Yang G и др. (2010) Повторное секвенирование генома 31 дикой и культивируемой сои выявило закономерности генетического разнообразия и селекции.Нат Жене
42: 1053–1059
10.1038/нг.715
[PubMed] [CrossRef] [Google Scholar] 27.
Hiremath PJ, Kumar A, Penmetsa RV, Farmer A, Schlueter JA, et al. (2012) Крупномасштабная разработка рентабельных анализов SNP-маркеров для оценки разнообразия и генетического картирования нута и сравнительного картирования бобовых. Завод Биотехнолог J
10: 716–732
10.1111/j.1467-7652.2012.00710.x
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]28.
Ганал М.В., Дурстевиц Г., Полли А., Берар А., Баклер Э.С. и др. (2011) Крупная кукуруза ( Zea mays L.) Матрица генотипирования SNP: развитие и генотипирование зародышевой плазмы, а также генетические гэпы для сравнения с эталонным геномом B73. ПЛОС ОДИН
6: e28334
10.1371/journal.pone.0028334
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]29.
Saxena RK, Penmetsa RV, Upadhyaya HD, Kumar A, Carrasquilla-Garcia N, et al. (2009) Крупномасштабная разработка экономичных анализов маркеров однонуклеотидного полиморфизма для генетического картирования голубиного гороха и сравнительного картирования бобовых. ДНК Res
19: 449–461
10.1093/dnares/dss025
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]30.Yu H, Xie W, Li J, Zhou F, Zhang Q (2014) Полногеномный массив SNP (RICE6K) для геномной селекции риса. Завод Биотехнолог J
12: 28–37
10.1111/пби.12113
[PubMed] [CrossRef] [Google Scholar] 31.
Ахунов Э., Николет С., Дворжак Дж. (2009) Генотипирование однонуклеотидного полиморфизма у полиплоидной пшеницы с помощью анализа Illumina GoldenGate. Теория Appl Genet
119: 507–517
10.1007/s00122-009-1059-5
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]32.
Гупта П.К., Рустги С., Мир Р.Р. (2008) Высокопроизводительные ДНК-маркеры на основе массива для улучшения урожая.Наследственность
101: 5–18
10.1038/hdy.2008.35
[PubMed] [CrossRef] [Google Scholar] 33.
Ганал М.В., Полли А., Гранер Э.М., Плиске Дж., Визеке Р. и др. (2012)Большие массивы SNP для генотипирования сельскохозяйственных растений. Джей Биоски
37: 821–828
10.1007/с12038-012-9225-3
[PubMed] [CrossRef] [Google Scholar] 34.
Бланка Дж. М., Паскуаль Л., Зиарсоло П., Нуэз Ф., Каньисарес Дж. (2011) Магистраль ngs: конвейер для очистки чтения, сопоставления и вызова snp с использованием последовательности следующего поколения. Геномика BMC
12: 285
10.1186/1471-2164-12-285
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]35.Deng X (2011) Seqgene: комплексное программное решение для анализа данных секвенирования экзома и транскриптома. БМК Биоинформатика
12: 267
10.1186/1471-2105-12-267
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]36.
Сана М.Э., Ясконе М., Марчетти Д., Палатини Дж., Галассо М. и др. (2011) Games идентифицирует и аннотирует мутации в проектах секвенирования следующего поколения. Биоинформатика
27: 9–13
10.1093/биоинформатика/btq603
[PubMed] [CrossRef] [Google Scholar] 37.
Qi J, Zhao F, Buboltz A, Schuster SC (2010) ingap: интегрированный конвейер анализа генома следующего поколения.Биоинформатика
26: 127–129
10.1093/биоинформатика/btp615
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]38.
ДеПристо М.А., Бэнкс Э., Поплин Р., Гаримелла К.В., Магуайр Дж.Р. и др. (2011)Структура для обнаружения вариаций и генотипирования с использованием данных секвенирования ДНК следующего поколения. Нат Жене
43: 491–498
10.1038/нг.806
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]39.
Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM (2010) Формат файла Sanger FASTQ для последовательностей с показателями качества и варианты Solexa/Illumina FASTQ.Нуклеиновые Кислоты Res
38: 1767–1771 гг.
10.1093/нар/гкп1137
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]42.
Li H, Ruan J, Durbin R (2008) Картирование коротких чтений секвенирования ДНК и вызов вариантов с использованием показателей качества картирования. Геном Res
18: 1851–1858 гг.
10.1101/гр.078212.108
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]45.
Li R, Yu C, Li Y, Lam T-W, Yiu S-M (2009) SOAP2: улучшенный сверхбыстрый инструмент для выравнивания коротких чтений. Биоинформатика
25: 1966–1967
10.1093/биоинформатика/btp336
[PubMed] [CrossRef] [Google Scholar]47.Ли Х., Хэндсакер Б., Высокер А., Феннелл Т., Руан Дж. и др. (2009) Формат выравнивания/карты последовательностей и SAMtools. Биоинформатика
25: 2078–2079 гг.
10.1093/биоинформатика/btp352
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]48.
Азам С., Такур В., Руперао П., Шах Т., Баладжи Дж. и др. (2012) Консенсусный вызов на основе охвата (CbCC) чтения коротких последовательностей и сравнение результатов CbCC для идентификации SNP в нуте ( Cicer arietinum ; Fabaceae ), культурных видах без эталонного генома.Ам Джей Бот
99: 186–192
10.3732/ajb.1100419
[PubMed] [CrossRef] [Google Scholar]50.
Li R, Li Y, Fang X, Yang H, Wang J и др. (2009)Обнаружение SNP для массивно-параллельного ресеквенирования всего генома. Геном Res
19: 1124–1132
10.1101/гр.088013.108
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]51.
Милн И., Байер М., Кардл Л., Шоу П., Стивен Г. (2010) Планшет — визуализация сборки последовательности нового поколения. Биоинформатика
26: 401–402
10.1093/биоинформатика/btp666
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]52.Милн И., Шоу П., Стивен Г., Байер М., Кардл Л. и др. (2010) Flapjack — графическая визуализация генотипа. Биоинформатика
26: 3133–3134
10.1093/биоинформатика/btq580
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]56.
Фличек П., Бирни Э. (2009) Смысл чтения последовательности: методы выравнивания и сборки. Нат Методы
6: 6–6
10.1038/н-мет.1376
[PubMed] [CrossRef] [Google Scholar]58.
Брэдбери П.Дж., Чжан З., Крун Д.Э., Касстевенс Т.М., Рамдосс Ю. и др. (2007) TASSEL: программное обеспечение для картирования ассоциаций сложных признаков в различных образцах.Биоинформатика
23: 2633–2635
10.1093/биоинформатика/btm308
[PubMed] [CrossRef] [Google Scholar]59.
Варшней Р.К., Сонг С., Саксена Р.К., Азам С., Ю С. и др. (2013) Черновая последовательность генома нута ( Cicer arietinum ) предоставляет ресурсы для улучшения признаков. Нат Биотехнолог
31: 240–246
10.1038/нбт.2491
[PubMed] [CrossRef] [Google Scholar] 60.
Фишер М., Снайдер Р., Пабингер С., Дандер А., Шоссиг А. и др. (2012) SIMPLEX: облачный конвейер для всестороннего анализа данных секвенирования экзома.ПЛОС ОДИН
7: e41948
10.1371/journal.pone.0041948
[бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
1093/нагрудник/bbp039
[PubMed] [CrossRef] [Google Scholar]2.
Варшней Р.К., Наяк С.Н., Мэй Г.Д., Джексон С.А. (2009)Технологии секвенирования следующего поколения и их значение для генетики и селекции сельскохозяйственных культур. Тенденции Биотехнологии
27: 522–530
10.1016/j.tibtech.2009.05.006
[PubMed] [CrossRef] [Google Scholar]3.
Маршалл Д.Дж., Хейворд А., Илз Д., Имельфорт М., Стиллер Дж. и др. (2010) Целевая идентификация геномных регионов с использованием TAGdb. Растительные методы
6: 19–19
10.1186/1746-4811-6-19
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]4.Майкл Т.П., Джексон С. (2013) Первые 50 геномов растений. Plant Gen 6: doi:10.3835/plantgenome2013.03.0001 [Google Scholar]5.
Дюран С., Эпплби Н., Варди М., Имельфорт М., Эдвардс Д. и др. (2009)Обнаружение полиморфизма одиночных нуклеотидов в ячмене с использованием autoSNPdb. Завод Биотехнолог J
7: 326–333
10.1111/j.1467-7652.2009.00407.x
[PubMed] [CrossRef] [Google Scholar]6.
Лу Т., Лу Г., Фан Д., Чжу С., Ли В. и др. (2010) Функциональная аннотация транскриптома риса при разрешении одного нуклеотида с помощью RNA-seq.Геном Res
20: 1238–1249 гг.
10.1101/гр.106120.110
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]7.
Дубей А., Фармер А., Шлютер Дж., Кэннон С.Б., Абернати Б. и др. (2011)Определение сборки транскриптома и ее использование для динамики генома и исследований профилирования транскриптома у голубиного гороха ( Cajanus cajan L.). ДНК Res
18: 153–164
10.1093/dnares/dsr007
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]8.
Гарг Р., Патель Р.К., Тьяги А.К., Джейн М. (2011) Сборка транскриптома нута De novo с использованием коротких прочтений для обнаружения генов и идентификации маркеров.ДНК Res
18: 53–63
10.1093/dnares/dsq028
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]9.
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, et al. (2011) Крупномасштабный транскриптомный анализ нута ( Cicer arietinum L.), бесхозной бобовой культуры полузасушливых тропиков Азии и Африки. Завод Биотехнолог J
9: 922–931
10.1111/j.1467-7652.2011.00625.x
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]10.
Дешам С., Ла Рота М., Раташак Дж. П., Биддл П., Турин Д. и др.(2010) Быстрое обнаружение однонуклеотидного полиморфизма по всему геному у сои и риса путем глубокого повторного секвенирования библиотек с уменьшенным представлением с помощью анализатора генома Illumina. Ген завода
3: 53–68
10.3835/геном растений2009.09.0026
[Перекрестная ссылка] [Академия Google] 11.
ван Тассел К.П., Смит Т.П.Л., Матукумалли Л.К., Тейлор Дж.Ф., Шнабель Р.Д. и соавт. (2008)Обнаружение SNP и оценка частоты аллелей путем глубокого секвенирования библиотек с уменьшенным представлением. Нат Методы
5: 247–252
10.1038/н-мет.1185
[PubMed] [CrossRef] [Google Scholar] 12.Гор М.А., Чиа Дж.М., Элшир Р.Дж., Сан К., Эрсоз Э.С. и др. (2009) Карта гаплотипов кукурузы первого поколения. Наука
326: 1115–1117
10.1126/наука.1177837
[PubMed] [CrossRef] [Google Scholar] 13.
Hyten DL, Cannon SB, Song Q, Weeks N, Fickus EW и др. (2010)Обнаружение SNP с высокой пропускной способностью посредством глубокого повторного секвенирования библиотеки с уменьшенным представлением для закрепления и ориентации каркасов в последовательности всего генома сои. Геномика BMC
11: 38–38
10.1186/1471-2164-11-38
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]15.van Orsouw NJ, Hogers RCJ, Janssen A, Yalcin F, Snoeijers S, et al. (2007) Снижение сложности полиморфных последовательностей (CRoPS): новый подход к обнаружению крупномасштабного полиморфизма в сложных геномах. PLoS один
2: e1172
10.1371/journal.pone.0001172
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]16.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, et al. (2008)Быстрое открытие SNP и генетическое картирование с использованием секвенированных маркеров RAD. PLoS один
3: e3376
10.1371/journal.pone.0003376
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]17.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, et al. (2011) Надежный подход простого генотипирования путем секвенирования (GBS) для видов с большим разнообразием. PLoS один
6: e19379
10.1371/journal.pone.0019379
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]18.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, et al. (2011)Полногеномное открытие генетических маркеров и генотипирование с использованием секвенирования следующего поколения. Нат Рев Жене
2: 499–510
10.1038/нрг3012
[PubMed] [CrossRef] [Google Scholar] 19.Huang X, Feng Q, Qian Q, Zhao Q, Wang L и др. (2009) Высокопроизводительное генотипирование путем полногеномного повторного секвенирования. Геном Res
19: 1068–1076
10.1101/гр.089516.108
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]20.
Мецкер М.Л. (2010) Технологии секвенирования — следующее поколение. Нат Рев Жене
11: 31–46
10.1038/nrg2626
[PubMed] [CrossRef] [Google Scholar] 22.
You FM, Huo N, Deal KR, Gu YQ, Luo MC и др. (2011)Обнаружение полногеномных SNP на основе аннотаций в большом и сложном геноме Aegilops tauschii с использованием секвенирования следующего поколения без эталонной последовательности генома.Геномика BMC
12: 59–59
10.1186/1471-2164-12-59
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]23.
Гаур Р., Азам С., Джина Дж., Хан А.В., Чоудхари С. и др. (2012)Обнаружение и генотипирование SNP с высокой пропускной способностью для построения карты насыщенного сцепления нута ( Cicer arietinum L.). ДНК Res
19: 357–373
10.1093/dnares/dss018
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]24.
Хаффорд М.Б., Сюй С., ван Хеерваарден Дж., Пихаярви Т., Чиа Дж.-М. и др. (2012) Сравнительная популяционная геномика одомашнивания и улучшения кукурузы.Нат Жене
44: 808–811
10.1038/нг.2309
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]25.
Xu X, Liu X, Ge S, Jensen JD, Hu F, et al. (2012) Повторное секвенирование 50 образцов культурного и дикого риса дает маркеры для идентификации агрономически важных генов. Нат Биотехнолог
30: 105–111
10.1038/нбт.2050
[PubMed] [CrossRef] [Google Scholar] 26.
Lam H-M, Xu X, Liu X, Chen W, Yang G и др. (2010) Повторное секвенирование генома 31 дикой и культивируемой сои выявило закономерности генетического разнообразия и селекции.Нат Жене
42: 1053–1059
10.1038/нг.715
[PubMed] [CrossRef] [Google Scholar] 27.
Hiremath PJ, Kumar A, Penmetsa RV, Farmer A, Schlueter JA, et al. (2012) Крупномасштабная разработка рентабельных анализов SNP-маркеров для оценки разнообразия и генетического картирования нута и сравнительного картирования бобовых. Завод Биотехнолог J
10: 716–732
10.1111/j.1467-7652.2012.00710.x
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]28.
Ганал М.В., Дурстевиц Г., Полли А., Берар А., Баклер Э.С. и др. (2011) Крупная кукуруза ( Zea mays L.) Матрица генотипирования SNP: развитие и генотипирование зародышевой плазмы, а также генетические гэпы для сравнения с эталонным геномом B73. ПЛОС ОДИН
6: e28334
10.1371/journal.pone.0028334
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]29.
Saxena RK, Penmetsa RV, Upadhyaya HD, Kumar A, Carrasquilla-Garcia N, et al. (2009) Крупномасштабная разработка экономичных анализов маркеров однонуклеотидного полиморфизма для генетического картирования голубиного гороха и сравнительного картирования бобовых. ДНК Res
19: 449–461
10.1093/dnares/dss025
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]30.Yu H, Xie W, Li J, Zhou F, Zhang Q (2014) Полногеномный массив SNP (RICE6K) для геномной селекции риса. Завод Биотехнолог J
12: 28–37
10.1111/пби.12113
[PubMed] [CrossRef] [Google Scholar] 31.
Ахунов Э., Николет С., Дворжак Дж. (2009) Генотипирование однонуклеотидного полиморфизма у полиплоидной пшеницы с помощью анализа Illumina GoldenGate. Теория Appl Genet
119: 507–517
10.1007/s00122-009-1059-5
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]32.
Гупта П.К., Рустги С., Мир Р.Р. (2008) Высокопроизводительные ДНК-маркеры на основе массива для улучшения урожая.Наследственность
101: 5–18
10.1038/hdy.2008.35
[PubMed] [CrossRef] [Google Scholar] 33.
Ганал М.В., Полли А., Гранер Э.М., Плиске Дж., Визеке Р. и др. (2012)Большие массивы SNP для генотипирования сельскохозяйственных растений. Джей Биоски
37: 821–828
10.1007/с12038-012-9225-3
[PubMed] [CrossRef] [Google Scholar] 34.
Бланка Дж. М., Паскуаль Л., Зиарсоло П., Нуэз Ф., Каньисарес Дж. (2011) Магистраль ngs: конвейер для очистки чтения, сопоставления и вызова snp с использованием последовательности следующего поколения. Геномика BMC
12: 285
10.1186/1471-2164-12-285
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]35.Deng X (2011) Seqgene: комплексное программное решение для анализа данных секвенирования экзома и транскриптома. БМК Биоинформатика
12: 267
10.1186/1471-2105-12-267
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]36.
Сана М.Э., Ясконе М., Марчетти Д., Палатини Дж., Галассо М. и др. (2011) Games идентифицирует и аннотирует мутации в проектах секвенирования следующего поколения. Биоинформатика
27: 9–13
10.1093/биоинформатика/btq603
[PubMed] [CrossRef] [Google Scholar] 37.
Qi J, Zhao F, Buboltz A, Schuster SC (2010) ingap: интегрированный конвейер анализа генома следующего поколения.Биоинформатика
26: 127–129
10.1093/биоинформатика/btp615
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]38.
ДеПристо М.А., Бэнкс Э., Поплин Р., Гаримелла К.В., Магуайр Дж.Р. и др. (2011)Структура для обнаружения вариаций и генотипирования с использованием данных секвенирования ДНК следующего поколения. Нат Жене
43: 491–498
10.1038/нг.806
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]39.
Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM (2010) Формат файла Sanger FASTQ для последовательностей с показателями качества и варианты Solexa/Illumina FASTQ.Нуклеиновые Кислоты Res
38: 1767–1771 гг.
10.1093/нар/гкп1137
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]42.
Li H, Ruan J, Durbin R (2008) Картирование коротких чтений секвенирования ДНК и вызов вариантов с использованием показателей качества картирования. Геном Res
18: 1851–1858 гг.
10.1101/гр.078212.108
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]45.
Li R, Yu C, Li Y, Lam T-W, Yiu S-M (2009) SOAP2: улучшенный сверхбыстрый инструмент для выравнивания коротких чтений. Биоинформатика
25: 1966–1967
10.1093/биоинформатика/btp336
[PubMed] [CrossRef] [Google Scholar]47.Ли Х., Хэндсакер Б., Высокер А., Феннелл Т., Руан Дж. и др. (2009) Формат выравнивания/карты последовательностей и SAMtools. Биоинформатика
25: 2078–2079 гг.
10.1093/биоинформатика/btp352
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]48.
Азам С., Такур В., Руперао П., Шах Т., Баладжи Дж. и др. (2012) Консенсусный вызов на основе охвата (CbCC) чтения коротких последовательностей и сравнение результатов CbCC для идентификации SNP в нуте ( Cicer arietinum ; Fabaceae ), культурных видах без эталонного генома.Ам Джей Бот
99: 186–192
10.3732/ajb.1100419
[PubMed] [CrossRef] [Google Scholar]50.
Li R, Li Y, Fang X, Yang H, Wang J и др. (2009)Обнаружение SNP для массивно-параллельного ресеквенирования всего генома. Геном Res
19: 1124–1132
10.1101/гр.088013.108
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]51.
Милн И., Байер М., Кардл Л., Шоу П., Стивен Г. (2010) Планшет — визуализация сборки последовательности нового поколения. Биоинформатика
26: 401–402
10.1093/биоинформатика/btp666
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]52.Милн И., Шоу П., Стивен Г., Байер М., Кардл Л. и др. (2010) Flapjack — графическая визуализация генотипа. Биоинформатика
26: 3133–3134
10.1093/биоинформатика/btq580
[Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]56.
Фличек П., Бирни Э. (2009) Смысл чтения последовательности: методы выравнивания и сборки. Нат Методы
6: 6–6
10.1038/н-мет.1376
[PubMed] [CrossRef] [Google Scholar]58.
Брэдбери П.Дж., Чжан З., Крун Д.Э., Касстевенс Т.М., Рамдосс Ю. и др. (2007) TASSEL: программное обеспечение для картирования ассоциаций сложных признаков в различных образцах.Биоинформатика
23: 2633–2635
10.1093/биоинформатика/btm308
[PubMed] [CrossRef] [Google Scholar]59.
Варшней Р.К., Сонг С., Саксена Р.К., Азам С., Ю С. и др. (2013) Черновая последовательность генома нута ( Cicer arietinum ) предоставляет ресурсы для улучшения признаков. Нат Биотехнолог
31: 240–246
10.1038/нбт.2491
[PubMed] [CrossRef] [Google Scholar] 60.
Фишер М., Снайдер Р., Пабингер С., Дандер А., Шоссиг А. и др. (2012) SIMPLEX: облачный конвейер для всестороннего анализа данных секвенирования экзома.ПЛОС ОДИН
7: e41948
10.1371/journal.pone.0041948
[бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]Низкое влияние различных панелей SNP из двух пайплайнов локусов зданий на популяционные геномные показатели RAD-Seq: тематическое исследование пяти различных водных видов | BMC Genomics
Куэйл М.А., Смит М., Коупленд П., Отто Т.Д., Харрис С.Р., Коннор Т.Р. и соавт. Рассказ о трех платформах для секвенирования следующего поколения: сравнение секвенаторов ion torrent, pacific biosciences и Illumina MiSeq. Геномика BMC.2012;13:341. https://doi.org/10.1186/1471-2164-13-341.
КАС Статья пабмед ПабМед Центральный Google ученый
Веттерстранд К.А. Стоимость секвенирования ДНК: Данные | НГРИ. 2020. https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data. По состоянию на 1 июля 2020 г.
Гудвин С., Макферсон Д.Д., МакКомби В.Р. Достижение совершеннолетия: десять лет технологий секвенирования следующего поколения. Нат Рев Жене.2016;17:333–51. https://doi.org/10.1038/nrg.2016.49.
КАС Статья пабмед ПабМед Центральный Google ученый
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Быстрое обнаружение SNP и генетическое картирование с использованием секвенированных маркеров RAD. ПЛОС Один. 2008;3(10):e3376. https://doi.org/10.1371/journal.pone.0003376.
КАС Статья пабмед ПабМед Центральный Google ученый
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML.Открытие полногеномных генетических маркеров и генотипирование с использованием секвенирования нового поколения. Нат Рев Жене. 2011;12:499–510. https://doi.org/10.1038/nrg3012.
КАС Статья пабмед Google ученый
Эндрюс К.Р., Гуд Дж.М., Миллер М.Р., Луикарт Г., Хоэнлоэ П.А. Использование возможностей RADseq для экологической и эволюционной геномики. Нат Рев Жене. 2016;17:81–92. https://doi.org/10.1038/nrg.2015.28.
КАС Статья пабмед ПабМед Центральный Google ученый
Ван С., Мейер Э., Маккей Дж. К., Мац М. В.2b-RAD: простой и гибкий метод полногеномного генотипирования. Нат Методы. 2012; 9: 808–10. https://doi.org/10.1038/nmeth.2023.
КАС Статья пабмед Google ученый
Барбанти А., Торрадо Х., Макферсон Э., Баргеллони Л., Франч Р., Каррерас С. и др. Помощь в принятии решений для надежного и экономичного секвенирования 2b-RAD и анализа генотипа у немодельных видов. Мол Эколь Ресурс. 2020; 20: 795–806. https://дои.орг/10.1111/1755-0998.13144.
КАС Статья Google ученый
О’Лири С.Дж., Пуриц Дж.Б., Уиллис С.К., Холленбек С.М., Портной Д.С. Это не те локусы, которые вы ищете: принципы эффективной фильтрации SNP для молекулярных экологов. Мол Экол. 2018;27:3193–206. https://doi.org/10.1111/mec.14792.
Артикул Google ученый
Диас-Арсе Н., Родригес-Эспелета Н.Выбор параметров анализа данных RAD-Seq для популяционной генетики: чем больше, тем лучше? Фронт Жене. 2019;10:533. https://doi.org/10.3389/fgene.2019.00533.
КАС Статья пабмед ПабМед Центральный Google ученый
Хубер М. Сборник двустворчатых моллюсков. Полноцветный справочник по 3300 морским двустворчатым моллюскам мира. Состояние двустворчатых моллюсков после 250 лет исследований. Хакенхайм: ConchBooks; 2010.
Google ученый
Фрике Р., Эшмайер В., Фонг Д.Д.CAS — каталог рыб Эшмейера — виды по семействам. 2020. http://researcharchive.calacademy.org/research/ichthyology/catalog/SpeciesByFamily.asp. По состоянию на 22 ноября 2020 г.
Google ученый
Левин Х.А., Робинсон Г.Э., Кресс В.Дж., Бейкер В.Дж., Коддингтон Дж., Крэндалл К.А. и др. Проект Earth BioGenome: секвенирование жизни для будущего жизни. Proc Natl Acad Sci. 2018; 115:4325–33. https://doi.org/10.1073/pnas.1720115115.
КАС Статья пабмед Google ученый
Рошетт, Северная Каролина, Катчен, Дж. М.Получение генотипов из данных короткого чтения RAD-seq с использованием стеков. Нат Проток. 2017;12:2640–59. https://doi.org/10.1038/nprot.2017.123.
КАС Статья пабмед Google ученый
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Стеки: набор инструментов для анализа популяционной геномики. Мол Экол. 2013;22(11):3124–40. https://doi.org/10.1111/mec.12354.
Артикул пабмед ПабМед Центральный Google ученый
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH.Стеки: построение и генотипирование локусов De Novo из коротких последовательностей. Г3. 2011;1(3):171–82. https://doi.org/10.1534/g3.111.000240.
КАС Статья пабмед Google ученый
Puritz JB, Hollenbeck CM, Gold JR. dDocent: конвейер вызова вариантов RADseq, разработанный для популяционной геномики немодельных организмов. Пир Дж. 2014;2:e431. https://doi.org/10.7717/peerj.431.
Артикул пабмед ПабМед Центральный Google ученый
Torkamaneh D, Laroche J, Bastien M, Abed A, Belzile F.Fast-GBS: новый конвейер для эффективного и высокоточного вызова SNP из данных генотипирования путем секвенирования. Биоинформатика BMC. 2017; 18:1–7. https://doi.org/10.1186/s12859-016-1431-9.
КАС Статья Google ученый
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, et al. TASSEL-GBS: высокопроизводительный конвейер для генотипирования путем секвенирования. ПЛОС Один. 2014;9(2):e
. https://doi.org/10.1371/журнал.поне.00
.
КАС Статья пабмед ПабМед Центральный Google ученый
Eaton DAR, Overcast I. ipyrad: интерактивная сборка и анализ наборов данных RADseq. Биоинформатика. 2020 г. https://doi.org/10.1093/bioinformatics/btz966.
Torkamaneh D, Laroche J, Belzile F. Вызов SNP для всего генома на основе данных генотипирования с помощью секвенирования (GBS): сравнение семи конвейеров и двух технологий секвенирования.ПЛОС Один. 2016;11(8):e0161333. https://doi.org/10.1371/journal.pone.0161333.
КАС Статья пабмед ПабМед Центральный Google ученый
Shafer ABA, Peart CR, Tusso S, Maayan I, Brelsford A, Wheat CW, et al. Биоинформационная обработка данных RAD-seq существенно влияет на генетический вывод нижестоящих популяций. Методы Экол Эвол. 2017; 8: 907–17. https://doi.org/10.1111/2041-210X.12700.
Артикул Google ученый
Пэрис Дж. Р., Стивенс Дж. Р., Катчен Дж. М.Затерянные в пространстве параметров: дорожная карта для стеков. Методы Экол Эвол. 2017; 8: 1360–73. https://doi.org/10.1111/2041-210X.12775.
Артикул Google ученый
Сович М.Г., Фрайс А.С., Гиббс Х.Л. AftrRAD: конвейер для точной и эффективной сборки данных RADseq de novo. Мол Эколь Ресурс. 2015;15:1163–71. https://doi.org/10.1111/1755-0998.12378.
КАС Статья пабмед Google ученый
Райт Б., Фаркухарсон К.А., Макленнан Э.А., Белов К., Хогг С.Дж., Грюбер К.Э.От эталонных геномов к популяционной геномике: сравнение трех конвейеров секвенирования с уменьшенным представлением, выровненных по эталонам, у двух видов диких животных. Геномика BMC. 2019;20:453. https://doi.org/10.1186/s12864-019-5806-y.
КАС Статья пабмед ПабМед Центральный Google ученый
Мартинес Л., Фрейре Р., Ариас-Перес А., Мендес Дж., Инсуа А. Модели генетической изменчивости в ареале распространения моллюсков Cerastoderma edule , выведенные из микросателлитов и митохондриальной ДНК.Мар биол. 2015; 162:1393–406. https://doi.org/10.1007/s00227-015-2676-y.
КАС Статья Google ученый
Вера М., Карлссон Дж., Эль Карлссон Дж., Кросс Т., Линч С., Камерманс П. и др. Текущий генетический статус, временная стабильность и структура остаточных популяций диких европейских плоских устриц: значение для сохранения и восстановления. Мар биол. 2016;163:239. https://doi.org/10.1007/s00227-016-3012-x.
Артикул Google ученый
Лейтвейн М., Гинан Б., Пузаду Дж., Демарэ Э., Берреби П., Ганьер П.А.Плотная кумжа ( Salmo trutta ) Карта сцепления показывает недавние хромосомные перестройки в роде Salmo и влияние отбора на связанное нейтральное разнообразие. Г3. 2017;7:1365–76. https://doi.org/10.1534/g3.116.038497.
Артикул пабмед Google ученый
Фергюсон А. Генетические различия среди кумжи, Salmo trutta , запасы и их значение для сохранения и управления видами.Свежая биол. 1989; 21:35–46.
Артикул Google ученый
Риос Н., Казанова А., Эрмида М., Пардо Б.Г., Мартинес П., Буза С. и др. Популяционная геномика Rhamdia quelen (Heptapteridae, siluriformes) выявляет глубокую дивергенцию и адаптацию в неотропическом регионе. Гены. 2020;11:109. https://doi.org/10.3390/genes11010109.
КАС Статья ПабМед Центральный Google ученый
Мануцци А., Зейн Л., Муньос-Мерида А., Гриффитс А.М., Вериссимо А.Популяционная геномика и филогеография донной прибрежной акулы ( Scyliorhinus canicula ) с использованием однонуклеотидных полиморфизмов 2b-RAD. Biol J Linn Soc. 2018; 126: 289–303. https://doi.org/10.1093/biolinnean/bly185.
Артикул Google ученый
Yan X, Nie H, Huo Z, Ding J, Li Z, Yan L и др. Последовательность генома моллюска проясняет молекулярную основу его бентической адаптации и необычайного разнообразия окраски раковины.iНаука. 2019;19:1225–37. https://doi.org/10.1016/j.isci.2019.08.049.
Артикул пабмед ПабМед Центральный Google ученый
Salmo trutta в сборе (NCBI). https://www.ncbi.nlm.nih.gov/assembly/GCF_1165.1. По состоянию на 26 июля 2020 г.
Ваджид Б., Серпедин Э. Руководство по сборке генома «Сделай сам». Краткая функциональная геномика. 2016; 15:1–9. https://doi.org/10.1093/bfgp/elu042.
КАС Статья пабмед Google ученый
Wang Y, Guo X. Хромосомная перестройка у пектиновых, выявленная с помощью локусов рРНК и значение для эволюции двустворчатых моллюсков. Биол Бык. 2004;207(3):247–56. https://doi.org/10.2307/1543213.
КАС Статья пабмед Google ученый
Такеучи Т., Коянаги Р., Гёджа Ф., Канда М., Хисата К., Фуджи М. и др.Экспансия генов, специфичных для двустворчатых моллюсков, в геноме жемчужной устрицы: последствия адаптации к сидячему образу жизни. Зоол Летт. 2016;2:3. https://doi.org/10.1186/s40851-016-0039-2.
Артикул Google ученый
Curole JP, Hedgecock D. Геномика двустворчатых моллюсков: осложнения, проблемы и перспективы на будущее. В: Лю Z (J), редактор. Геномные технологии аквакультуры. Оксфорд: Blackwell Publishing Ltd; 2007. с. 525–43.
Глава Google ученый
Паскье Дж., Кабау С., Нгуен Т., Жуанно Э., Северак Д., Брааш И. и др.Эволюция генов и экспрессия генов после дупликации всего генома у рыб: база данных PhyloFish. Геномика BMC. 2016;17:368. https://doi.org/10.1186/s12864-016-2709-z.
КАС Статья пабмед ПабМед Центральный Google ученый
Macqueen DJ, Johnston IA. Хорошо ограниченная оценка времени дупликации всего генома лососевых выявляет значительное отделение от видовой диверсификации. Proc R Soc B Biol Sci.2014; 281:1778. https://doi.org/10.1098/rspb.2013.2881.
Артикул Google ученый
Бертело С., Брюне Ф., Шалопин Д., Хуанчич А., Бернар М., Ноэль Б. и др. Геном радужной форели дает новое представление об эволюции после дупликации всего генома у позвоночных. Нац коммун. 2014;5:2. https://doi.org/10.1038/ncomms4657.
Артикул Google ученый
Donoghue PCJ, Purnell MA.Дублирование генома, вымирание и эволюция позвоночных. Тенденции Экол Эвол. 2005;20(6):312–9. https://doi.org/10.1016/j.tree.2005.04.008.
Артикул пабмед Google ученый
Benestan LM, Ferchaud AL, Hohenlohe PA, Garner BA, Naylor GJP, Baums IB, et al. Сохранение геномики естественных и управляемых популяций: построение концептуальной и практической основы. Мол Экол. 2016;25:2967–77. https://doi.org/10.1111/mec.13647.
Артикул пабмед Google ученый
Hendricks S, Anderson EC, Antao T, Bernatchez L, Forester BR, Garner B, et al. Последние достижения в области сохранения и анализа данных популяционной геномики. Приложение Эвол. 2018;11:1197–211. https://doi.org/10.1111/eva.12659.
Артикул ПабМед Центральный Google ученый
Hodel RGJ, Chen S, Payton AC, McDaniel SF, Soltis P, Soltis DE.Добавление локусов улучшает филогеографическое разрешение в красных мангровых зарослях, несмотря на увеличение пропущенных данных: сравнение микросателлитов и RAD-Seq и исследование фильтрации локусов. Научный доклад 2017; 7: 17598. https://doi.org/10.1038/s41598-017-16810-7.
КАС Статья пабмед ПабМед Центральный Google ученый
Мастретта-Янес А., Арриго Н., Альварес Н., Йоргенсен Т.Х., Пиньеро Д., Эмерсон Б.К. Секвенирование ДНК, связанное с сайтом рестрикции, оценка ошибки генотипирования и оптимизация сборки de novo для генетического вывода популяции.Мол Эколь Ресурс. 2015;15:28–41. https://doi.org/10.1111/1755-0998.12291.
КАС Статья пабмед Google ученый
Fountain ED, Pauli JN, Reid BN, Palsbøll PJ, Peery MZ. Поиск правильного охвата: влияние охвата и качества последовательности на частоту ошибок генотипирования полиморфизма одиночных нуклеотидов. Мол Эколь Ресурс. 2016; 16: 966–78. https://doi.org/10.1111/1755-0998.12519.
КАС Статья пабмед Google ученый
Милан М., Марозо Ф., Далла Ровере Г., Карраро Л., Феррарессо С., Патарнелло Т. и др.Отслеживание морепродуктов с высоким пространственным разрешением с использованием данных NGS и машинного обучения: сравнение микробиома и SNP. Пищевая хим. 2019; 286: 413–20. https://doi.org/10.1016/j.foodchem.2019.02.037.
КАС Статья пабмед Google ученый
Марозо Ф., Де Грасия К.П., Иглесиас Д., Као А., Диас С., Вильяльба А. и др. Полезная панель SNP для различения двух видов моллюсков, Cerastoderma edule и C. glaucum , совместно встречающихся в некоторых европейских слоях, и их предполагаемых гибридов.Гены. 2019;10:760. https://doi.org/10.3390/genes10100760.
КАС Статья ПабМед Центральный Google ученый
Bouza C, Castro J, Sánchez L, Martinez P. Аллозимные доказательства парапатрической дифференциации кумжи ( Salmo trutta L .) в бассейне атлантической реки Пиренейского полуострова. Мол Экол. 2001; 10:1455–69. https://doi.org/10.1046/j.1365-294X.2001.01272.x.
КАС Статья пабмед Google ученый
Вера М., Корти М., Санс Н., Гарсия-Марин Х.Л.Поддержание эндемичной линии кумжи ( Salmo trutta ) в бассейне реки Дуэро. J Zool Syst Evol Res. 2010;48:181–7. https://doi.org/10.1111/j.1439-0469.2009.00547.x.
Артикул Google ученый
Мартинес П., Боуза С., Кастро Дж., Эрмида М., Пардо Б.Г., Санчес Л. Анализ вторичного контакта между расходящимися линиями кумжи Salmo trutta L. из бассейна Дуэро с использованием микросателлитов и ПДРФ мтДНК.Дж. Фиш Биол. 2007; 71: 195–213. https://doi.org/10.1111/j.1095-8649.2007.01551.x.
КАС Статья Google ученый
Perdices A, Bermingham E, Montilla A, Doadrio I. Эволюционная история рода Rhamdia (Teleostei: Pimelodidae) в Центральной Америке. Мол Филогенет Эвол. 2002; 25: 172–89. https://doi.org/10.1016/S1055-7903(02)00224-5.
КАС Статья пабмед Google ученый
Руссе Ф.GENEPOP’007: полная переработка программного обеспечения GENEPOP для Windows и Linux. Мол Эколь Ресурс. 2008; 8: 103–6. https://doi.org/10.1111/j.1471-8286.2007.01931.x.
Артикул пабмед Google ученый
Притчард Дж. К., Стивенс М., Доннелли П. Вывод о структуре популяции с использованием данных о многолокусных генотипах. Генетика. 2000; 155:945–59.
КАС пабмед ПабМед Центральный Google ученый
Фу Л., Ню Б., Чжу З., Ву С., Ли В.CD-HIT: ускорено для кластеризации данных секвенирования нового поколения. Биоинформатика. 2012;28:3150–2. https://doi.org/10.1093/bioinformatics/bts565.
КАС Статья пабмед ПабМед Центральный Google ученый
Ли В., Годзик А. Cd-hit: быстрая программа для кластеризации и сравнения больших наборов последовательностей белков или нуклеотидов. Биоинформатика. 2006; 22:1658–9. https://doi.org/10.1093/bioinformatics/btl158.
КАС Статья Google ученый
Лангмид Б., Трапнелл С., Поп М., Зальцберг С.Л.Сверхбыстрое и эффективное с точки зрения памяти выравнивание коротких последовательностей ДНК с геномом человека. Геном биол. 2009;10:R25.
Артикул Google ученый
Bolger AM, Lohse M, Usadel B. Trimmomatic: гибкий триммер для данных последовательностей Illumina. Биоинформатика. 2014;30:2114–20. https://doi.org/10.1093/bioinformatics/btu170.
КАС Статья пабмед ПабМед Центральный Google ученый
Эндрюс С.FastQC: инструмент контроля качества для высокопроизводительных данных последовательностей. 2010. Доступно в Интернете по адресу: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Google ученый
Lischer HE, Excoffier L. PGDSpider: инструмент автоматического преобразования данных для соединения программ популяционной генетики и геномики. Биоинформатика. 2012; 28: 298–9. https://doi.org/10.1093/bioinformatics/btr642.
КАС Статья пабмед Google ученый
Кинан К., Макгиннити П., Кросс Т.Ф., Крозье В.В., Продёль П.А.DiveRsity: пакет R для оценки и исследования параметров популяционной генетики и связанных с ними ошибок. Методы Экол Эвол. 2013; 4: 782–8. https://doi.org/10.1111/2041-210X.12067.
Артикул Google ученый
Бенье Ф., Гловер К.А. ParallelStructure: пакет R для распространения параллельных запусков программы популяционной генетики STRUCTURE на многоядерных компьютерах. ПЛОС Один. 2013;8(7):e70651. https://doi.org/10.1371/journal.pone.0070651.
КАС Статья пабмед ПабМед Центральный Google ученый
Эрл Д.А. фон Холдт БМ. STRUCTURE HARVESTER: веб-сайт и программа для визуализации выходных данных STRUCTURE и реализации метода Эванно. Сохраните ресурсы Genet. 2012;4:359–61. https://doi.org/10.1007/s12686-011-9548-7.
Артикул Google ученый
Эванно Г., Регнаут С., Гуде Дж.Определение количества скоплений особей с помощью программного обеспечения СТРУКТУРА: имитационное исследование. Мол Экол. 2005; 14: 2611–20. https://doi.org/10.1111/j.1365-294X.2005.02553.x.
КАС Статья Google ученый
Копельман Н.М., Майзель Дж., Якобссон М., Розенберг Н.А., Мэйроуз И. Клампак: программа для определения режимов кластеризации и выводов о структуре популяции в K. Mol Ecol Resour. 2015;15:1179–91. https://дои.орг/10.1111/1755-0998.12387.
КАС Статья пабмед ПабМед Центральный Google ученый
Джомбарт Т. Адегенет: R-пакет для многомерного анализа генетических маркеров. Биоинформатика. 2008;24(11):1403–5. https://doi.org/10.1093/bioinformatics/btn129.
КАС Статья пабмед Google ученый
Джомбарт Т., Ахмед И. Адегенет 1.3-1: новые инструменты для анализа полногеномных данных SNP. Биоинформатика. 2011;27(21):3070–1. https://doi.org/10.1093/bioinformatics/btr521.
КАС Статья пабмед ПабМед Центральный Google ученый
Foll M, Gaggiotti O. Метод сканирования генома для идентификации выбранных локусов, подходящих как для доминантных, так и для кодоминантных маркеров: байесовская перспектива. Генетика. 2008; 180:977–93. https://doi.org/10.1534/genetics.108.0.
Артикул пабмед ПабМед Центральный Google ученый
Точность и эффективность конвейеров вызова вариантов зародышевой линии для данных генома человека
В этом исследовании мы эмпирически оценили производительность различных конвейеров (и их комбинаций) для вызова вариантов зародышевой линии с использованием реальных и смоделированных данных WGS. Наши результаты показали, что DeepVariant ( DV_dragen3 или DV_gatk4 ) показывает более высокую точность в вызовах SNP для одного набора данных NA12878 (SRR6794144) и двух «синтетически-диплоидных» наборов данных, а также в вызовах indel для двух наборов данных NA12878.Несмотря на лучшую производительность, баллы F1, полученные в сравнительной оценке NA12878, были ниже, чем результаты, опубликованные в FDA Truth Challenge: 0,9912–0,9959 по сравнению с 0,9996 (верхний уровень pFDA) для вызовов SNP и 0,9897–0,9717 по сравнению с 0,9934 (верхний уровень pFDA) для indel. звонки. Это изменение, вероятно, является результатом различий в процедуре сравнительного анализа теста pFDA Truth, в котором образец NA12878 использовался для обучения, а образец HG002 использовался для тестирования. Наилучшие результаты сравнительного анализа в тесте pFDA Truth Challenge были получены из сравнения HG002.Точность конвейера DRAGEN ( Dragen3_raw ) дала лучшую производительность как в вызовах SNP, так и в вызовах indel для смоделированного набора данных, а также в вызовах indel для «синтетически-диплоидных» наборов данных, несмотря на то, что он не достиг таких высоких показателей F1-показателя, как DeepVariant. в тесте набора данных NA12878. На самом деле различия в оценках бенчмаркинга между DRAGEN и DeepVariant довольно малы (рис. 2 и рис. S1). В частности, анализ стратификации консервативных и кодирующих областей предполагает почти одинаковую точность между ними.Таким образом, слияние вариантов, вызываемых несколькими конвейерами, может уменьшить количество ложноотрицательных результатов в сравнительном исследовании, что потенциально положительно влияет на оценку F1. Однако это приведет к ложным срабатываниям, в частности, для неконгруэнтных генотипов, фазированных разными вызывающими. С точки зрения компромиссного соотношения между отзывом и точностью, оценка F1 не всегда указывает на улучшение (она зависит от соотношения между уменьшением числа ложноотрицательных результатов и усилением ложноположительных результатов).
Наиболее важным преимуществом платформы DRAGEN является время вычислений и, следовательно, пропускная способность при обработке огромных объемов данных.Действительно, в этом исследовании эффективность работы платформы DRAGEN была намного выше, чем у GATK и DeepVariant, благодаря поддержке аппаратных ускорений. Основываясь на этих соображениях и измеренных нами результатах точности, представляется разумным рекомендовать использовать конвейер DRAGEN отдельно ( Dragen3_raw ) или в комбинации ( DV_Dragen3 ), где DRAGEN используется для восходящей обработки, а DeepVariant — для обработки. последующая обработка, чтобы получить баланс точности и эффективности для вызова вариантов зародышевой линии из данных WGS.
Хотя в этом исследовании платформа DRAGEN показала наилучшие показатели эффективности работы, реальное время выполнения на кластерах HPC никогда не достигало производительности, заявленной производителем. Даже когда бенчмаркинг проводился на локальной виртуальной машине, где более быстрый ввод-вывод в локальной файловой системе ext4 мог повысить скорость работы по сравнению с сетевой файловой системой BeeGFS в кластере HPC, можно было наблюдать лишь незначительные улучшения в потреблении времени. (Рисунок S5). Таким образом, еще есть возможности для оптимизации среды выполнения платформы DRAGEN в отношении ее реализации на уровне инфраструктуры и оборудования.По сравнению с DRAGEN оптимизация эффективности работы для GATK и DeepVariant не была достигнута в вычислительной среде нашего исследования. Например, DeepVariant может получить ускорение в 2,5 раза, используя высокопроизводительный графический процессор, поскольку его алгоритм вызова вариантов основан на анализе изображений. Для GATK геном был разделен на 14 фракций по хромосомам, скаффолдам и контигам, и они были запущены в стратегии «разброс-сбор». В кластере HPC было 64 ядра на узел, поэтому геном в идеале мог быть разделен на такое же количество делений, как и количество ядер, и запускаться параллельно.Несмотря на эти оптимизации, ни DeepVariant, ни GATK не смогли бы достичь эффективности DRAGEN, поскольку для них не было разработано аппаратно-ускоренных реализаций алгоритмов геномного анализа.
Для оценки производительности вызывающих вариантов зародышевой линии с использованием реальных данных применялись два типа наборов эталонных вызовов истинности с высокой достоверностью: эталонные данные GiaB (образец NA12878) и «синтетически-диплоидная» смесь двух гаплоидных клеточных линий. Следует учитывать построение набора истинности, а также сильных и слабых сторон на основе типа варианта и контекста генома.Наборы контрольных показателей GiaB были созданы на основе консенсуса нескольких инициаторов вариантов в отношении короткого секвенирования Illumina с помощью анализа родословной, интеграции структурных вариантов, идентифицированных с помощью технологий длинных фрагментов PacBio и 10X Genomics, и анализа генома HuRef с использованием секвенирования по Сэнгеру . 39 . Почти все «настоящие» варианты в образце NA12878 присутствуют в файлах ресурсов (например, dbSNP, 1000 геномов и обучающих данных для DeepVariant), используемых для запуска конвейера.В этом случае результаты, вероятно, являются переобученными, поскольку ответ использовался все время. Кроме того, набор истинных вызовов NA12878 исключает более сложные типы вариантов в области с умеренно дивергированными повторами и сегментарными дупликациями, поскольку консенсус в таких областях не достигнут. Это приведет к смещению наборов данных GiaB в сторону областей генома, которые «легко секвенировать и анализировать».
Настоящий «синтетически-диплоидный» набор вызовов был создан путем сборки длинных ридов, секвенированных из двух гаплоидных клеточных линий (CHM1 и CHM13) с использованием технологии PacBio.Это можно считать заслуживающим доверия, так как не существует гетерозиготных сайтов, способных запутать сборку. Эксклюзивное использование PacBio без учета недостатков, порожденных технологией короткого считывания Illumina, обеспечивает меньшую корреляцию между режимами отказа этого метода в данных короткого считывания и доверительных областях. Это позволяет проводить бенчмаркинг в регионах, которые трудно отобразить с помощью коротких чтений. Однако «синтетически-диплоидный» набор вызовов в настоящее время содержит некоторые ошибки, которые изначально присутствовали в длинных чтениях 27 .Таким образом, для сравнений 27,39 рекомендуется использовать менее строгую стратегию сравнительного анализа (метод локальных совпадений). Здесь также была проведена оценка с использованием «совпадения генотипа», применяемого в наборах данных NA12878 (таблица S4). Что касается показателей SNP, DeepVariant ( DV_gatk4 или DV_dragen3) неизменно оценивался как лучший в соответствии с их соответствующими баллами F1. Что касается показателей производительности вызовов indel, Dragen3_raw и GATK4_raw имели лучшее значение для наборов данных ERR1341793 и ERR1341796 соответственно.Как и ожидалось, полнота, точность и оценка F1 вставок относительно низки по сравнению с показателями, полученными методом «локального сопоставления». Точная оценка точности генотипов по точным изменениям последовательности в полях REF и ALT файла VCF для сравнительного анализа «синтетических-диплоидных» данных остается сложной задачей. Следовательно, требуется менее строгая методология, такая как подход «местного сопоставления». Одним из преимуществ является устойчивость к репрезентативным различиям вариантов в наборах истинности и запроса.В целом, характеристики этих двух наборов данных об истинности делают их очень ценными для проведения комплексной сравнительной оценки различных инструментов биоинформатики.
В дополнение к реальным данным WGS мы создали два смоделированных набора данных WGS на основе случайного и заданного пользователем профиля мутации. Одним из преимуществ использования данных, смоделированных in silico, для сравнительного анализа является то, что известны все «истинно» положительные SNP и вставки без наличия противоречивых генотипов. Расчет F1-показателя является более точным из-за сниженного риска переоценки ложноотрицательных результатов.Кроме того, в смоделированных данных покрытие считывания по всему региону генома имеет более равномерное распределение, чем в реальных данных, поэтому количество вариантов ошибок вызова, возникающих из-за низкого охвата в некоторых регионах, может быть уменьшено. С другой стороны, точность поиска вариантов в смоделированных данных легко достигает насыщения (рис. S1), поскольку смоделированные данные могут достичь идеального соответствия (почти 100%, таблица S1) эталонному геному, что дает преимущества при вызове вариантов как для SNP, так и для вставки Кроме того, была обнаружена разница в анализе стратификации содержания GC между данными in silico и реальными данными, при этом показатели эффективности, показанные в смоделированных данных, меньше расходятся (рис. S3).Точно так же как ложноположительные, так и отрицательные варианты, вызываемые эталонными конвейерами в смоделированных данных, не зависят от каких-либо типов смещений SNP в распределении сигнатур замещения (рис. S6). Все эти систематические расхождения между смоделированными и реальными данными предполагают, что данные in silico не могут отразить истинную экспериментальную изменчивость и всегда менее сложны, чем реальные данные 41,42 . В частности, модели, используемые для моделирования данных, могут не воспроизводить идентичную сложность последовательности в реальных данных в отношении всех биологических и технологических особенностей.Например, некоторые важные параметры моделирования, такие как ПЦР-амплификация во время подготовки библиотеки, погрешность охвата GC%, ошибки секвенирования и профиль мутаций, были эмпирически изучены из выбранных известных наборов данных без учета специфичности и разнообразия образцов в целом. Как показали результаты, протестированные конвейеры могут хорошо идентифицировать большинство истинных срабатываний, не вводя переменных ложных срабатываний, когда вызов вариантов выполняется при смоделированных чтениях. Хотя модели не полностью соответствуют реальному сценарию, моделирование по-прежнему является важным подходом для сравнительной оценки различных конвейеров биоинформатики с аналогичной функциональностью.Однако следует отметить, что применение смоделированных данных в бенчмаркинге может только дополнять реальные экспериментальные данные золотого стандарта в качестве полезного дополнения для тестирования и разработки вычислительных инструментов. Данные in silico не заменяют использование физических стандартов, которые измеряют весь диапазон вариаций, встречающихся в клинической диагностике 42 .
В передовых методах GATK и DRAGEN настоятельно рекомендуется применять повторную калибровку показателя качества вариантов (VQSR) для фильтрации необработанных вызовов SNP и indel, сгенерированных HaplotypeCaller, и для удаления артефактов вызова.Теоретически VQSR уравновешивает чувствительность и специфичность во время фильтрации вариантов. Однако оценка F1 была ниже как в реальных, так и в смоделированных данных, за исключением Dragen3_vqsr в NA12878_SRR679414 после фильтрации VQSR, хотя точность достигла наивысшего значения. На рис. 2 показатели точности в среднем выросли только на 0,15 % и 0,5 % для SNP и вставок соответственно, в то время как полнота пострадала от более значительного падения, что является значительным для GATK4_vqsr (например, снижение на 3 % для SNP и 4% для вставок в наборе данных NA12878_PrecisionFDA).Следовательно, рассчитанная оценка F1 не показала ожидаемого улучшения. Потенциально это можно объяснить тем фактом, что VQSR выполнялся на одном образце за раз, что приводило к нестабильности из-за сбоя сходимости моделирования основного алгоритма. Это может привести к необходимости довольно «строгих» критериев при фильтрации исходных вызовов вариантов и привести к более низкому значению отзыва. Кроме того, мы столкнулись с некоторыми проблемами при выполнении анализа VQSR на смоделированных данных WGS с параметрами по умолчанию, поскольку было недостаточно вариантов для обучения как значимого «плохого набора» для эффективной дискриминации кластеров.Вместо этого мы уменьшили количество параметров максимального гаусса до 2 для делеций и 4 для SNP и заставили программу сгруппировать варианты в меньшее количество кластеров, чтобы удовлетворить статистические требования. В целом, наши результаты показывают, что нет необходимости выполнять контроль VQSR для анализа одного образца, и на самом деле необработанные нефильтрованные файлы VCF имеют хороший баланс между отзывом и точностью для GATK и DRAGEN.
Необходимо упомянуть несколько предостережений и ограничений текущего исследования. Во-первых, варианты вызова выполнялись конвейерами с использованием их параметров по умолчанию.Было бы интересно попытаться оптимизировать параметры и настройки для каждого конвейера, что потенциально может повысить точность вызова вариантов. Однако в целом это трудоемкий процесс, иногда требующий общения с авторами каждого инструмента для глубокого изучения использования параметров. Во-вторых, мы провели сравнительное исследование, используя как реальные, так и смоделированные данные. Еще один метод заключается в разработке «полусимуляционных» наборов данных, которые объединяют реальные экспериментальные данные с данными in silico (т.е. вычислительный) импульсный сигнал. Например, путем объединения ячеек из «нулевых» (например, здоровых) образцов с подмножеством ячеек из образцов, которые, как ожидается, содержат истинный дифференциальный сигнал. Эта стратегия может создавать наборы данных с более реалистичными уровнями изменчивости и корреляции, а также с достоверностью данных. Наконец, мы не включили все доступные варианты зародышевой линии, вызывающие конвейеры для сравнительного исследования, и для этого исследования были выбраны три из них (т. е. GATK, DRAGEN и DeepVariant), хотя существуют и другие с аналогичной функциональностью (например,грамм. Стрелка2). Мы сосредоточились на этих трех, потому что они представляют собой самые современные и широко используемые инструменты для вызова вариантов зародышевой линии с использованием данных WGS. Недавно команда GATK объявила о сотрудничестве с командой Illumina DRAGEN для совместной разработки методов анализа и конвейеров для вызова вариантов с коротким чтением. Вероятно, в ближайшем будущем будет выпущен DRAGEN-GATK, который, по-видимому, сможет предоставить исследователям быстрые, воспроизводимые и точные инструменты в среде с открытым исходным кодом и заслуживает внимания в дальнейших исследованиях.
В заключение, наш бенчмаркинг на реальных и смоделированных наборах данных WGS показывает, что конвейеры DRAGEN и DeepVariant обладают высокой точностью при вызове небольших зародышевых вариантов, и нет существенных различий в их характеристиках F1. Платформа DRAGEN показала превосходные результаты в сверхбыстром анализе данных WGS для обнаружения SNP и indel и, следовательно, имеет большой потенциал для внедрения в рутинную геномную медицину, где скорость может иметь решающее значение. Комбинация конвейеров DeepVariant и DRAGEN также может предложить быстрый, эффективный и надежный способ крупномасштабного анализа данных WGS и пройти долгий путь к надежному и последовательному вызову вариантов при преобразовании информации о генетических вариантах в медицинскую диагностику.
В 2020 году газовые компании будут меньше устранять утечки благодаря совершенствованию технологии мониторинга
Мобильное обнаружение утечек получило развитие в газовых компаниях, и эта тенденция ежегодно влияет на деятельность по устранению утечек. |
Устранение утечек крупными газовыми компаниями США в 2020 году сократилось на 6,6% по сравнению с 2019 годом, поскольку компании воспользовались преимуществами более совершенных технологий и более раннего ремонта и замены трубопроводов.
Количество ремонтов сократилось до 457 838, согласно анализу S&P Global Market Intelligence, основанному на федеральных данных коммунальных предприятий, работающих на природном газе, имеющих не менее 5000 миль распределительных сетей и линий обслуживания. Это было первое годовое падение ремонтной активности группы, зафиксированное в доступных данных с 2017 года.
Например, компания CenterPoint Energy Resources Corp., устранившая наибольшее количество утечек в 2020 году и эксплуатирующая самую крупную систему в группе, столкнулась с сокращением ремонтных работ с 58 087 утечек в 2019 году до 56 392 утечек в 2020 году, т. е. 2.9% скидка. Компания заявила, что значительный вклад, вероятно, внес ее опыт использования технологии обнаружения утечек Picarro Inc., устанавливаемой на транспортном средстве. По словам представителя CenterPoint Energy Inc. Росса Корсона, поскольку этот метод более эффективен и чувствителен, чем традиционные методы обнаружения, CenterPoint выявил больше утечек в ходе первого цикла исследований в последние годы. По мере того, как компания завершает дополнительные циклы обследования, она рассчитывает выявить меньше утечек из-за всплеска активности по устранению утечек в первом цикле, пояснил Корсон.
Данные подтверждают это. CenterPoint сообщил об увеличении количества ремонтов утечек в 2019 году на 11,4%. В 2020 году, когда прошел второй цикл исследований Picarro, количество ремонтов сократилось на 2,9%. Данные охватывают присутствие CenterPoint в Арканзасе, Луизиане, Миннесоте, Миссисипи, Оклахоме и Техасе. Недавно компания развернула технологию Picarro на своих территориях обслуживания в Огайо и Индиане, которые она приобрела в 2019 году.
Было неясно, в какой степени блокировки COVID-19 сыграли роль в снижении ремонтной активности.Газовые коммунальные предприятия в целом сообщали, что в 2020 году работа по обеспечению целостности системы продолжалась высокими темпами, чему способствовало назначение федеральным правительством сотрудников коммунальных служб в качестве основных работников и усилия отрасли на уровне штата и на местном уровне по обеспечению доступа к территориям обслуживания.
Не вызывает сомнений то, что десятилетнее давление на газовые компании со стороны правительства и общественности с целью устранения утечек вынудило крупные коммунальные предприятия усилить обнаружение утечек. Общеотраслевой переход на пластиковые трубы из старых материалов помог сократить количество утечек газа, наряду с резким ростом ремонтных работ, вызванным внедрением новой технологии обнаружения.
Чтобы повысить безопасность и сократить выбросы, коммунальные предприятия «усердно работали над улучшением того, как они используют новые инструменты и технологии для выявления небольших утечек», — сказал Эндрю Лу, вице-президент по эксплуатации и инжинирингу Американской газовой ассоциации. По словам Лу, коммунальные службы предприняли «более агрессивные шаги по устранению и устранению таких небольших утечек с меньшей концентрацией».
Седьмой год подряд Consolidated Edison Co. of New York Inc.показал самый высокий коэффициент устранения утечек. Но второй год подряд компания наблюдает, как ее соотношение и общее количество устраненных утечек снижаются до самого низкого уровня с тех пор, как Market Intelligence начала анализировать данные в 2013 году. -падение по сравнению с прошлым годом в 2019 году.
ConEdison частично объяснил это снижение зрелой программой замены чугунных и незащищенных стальных труб, на которые приходилось 12,2% и 15,6% от общего пробега на конец 2020 года соответственно.Тем не менее, компания также увидела, что ущерб, нанесенный подрядчиком ее системе, упал до самого низкого уровня за всю историю, поскольку компания развернула прогнозную аналитику для уменьшения непреднамеренного ущерба, говорится в электронном письме ConEdison.
Рейтинг устранения утечек в потоке
Анализ Market Intelligence показал, что на 23 коммунальных предприятия, которые по отдельности устранили более 5000 утечек, приходится 71% ремонтных работ, когда их усилия суммируются, что примерно соответствует уровню предыдущего года. Как и в прошлые годы, группа, наиболее активная в устранении утечек, состояла в основном из коммунальных служб, которые эксплуатируют старые системы с неизолированными стальными и чугунными трубами, подверженными утечкам.
Для составления рейтинга Market Intelligence рассчитала коэффициент устранения утечек для каждой компании, который измерял общий объем ремонта утечек компании, согласно данным Управления по безопасности трубопроводов и опасных материалов США, по сравнению с общим пробегом распределительных магистральных и сервисных линий компании.
Поскольку коммунальные предприятия обычно разбивают работу по обеспечению целостности системы на несколько лет, чтобы смягчить последствия выставления счетов для клиентов, рейтинги остаются относительно стабильными из года в год.Такие факторы, как слияния и поглощения, могут привести к большим сдвигам, особенно когда газовая компания приобретает или продает распределительную систему с большим количеством подверженных утечкам труб.
Данные за 2020 год и комментарии компаний свидетельствуют о том, что внедрение процессов и технологий также потрясло рейтинги в последние годы и может продолжать делать это в будущем.
Компания Peoples Gas Light and Coke Co. сообщила об увеличении объема работ по устранению утечек в 2020 году на 30,1%. WEC Energy Group Inc.дочерняя компания, которая распределяет газ в Чикаго, заявила, что постоянные усилия по улучшению управления данными об утечках привели к тому, что в 2020 году Peoples зарегистрировала большее количество устранений утечек. WEC Energy реализовала программу модернизации своей системы распределения природного газа в дополнение к ремонту. утечки, написал в электронном письме пресс-секретарь Брендан Конвей. Компания Peoples Gas Light резко поднялась в рейтинге 2020 года, заняв 13-е место по коэффициенту устранения утечек по сравнению с 31-й позицией в 2019 году.
При этом Peoples нарушила общую тенденцию 2020 года.Только четыре из 20 ведущих компаний, ранжированных по показателю устранения утечек, сообщили об увеличении объемов устранения утечек, причем в двух случаях рост был близок к неизменному.
Формирование будущего
Усовершенствования и повышение эффективности могут помочь коммунальной отрасли, поскольку законодатели и регулирующие органы предъявляют новые требования к операторам инфраструктуры по предотвращению утечек метана, согревающего планету, в попытке смягчить последствия изменения климата. Демократы в Конгрессе выдвинули плату за метан в рамках пакета согласования бюджета.
Газовые коммунальные предприятия с самым низким коэффициентом устранения утечек обычно используют системы с высоким процентным содержанием современных пластиковых магистралей и незначительным количеством чугунных и голых стальных труб. В дополнение к тому, что пластиковые активы менее подвержены утечкам, они могут окупиться, поскольку промышленность предпринимает шаги по смешиванию низкоуглеродистого водорода в своих системах для их обезуглероживания.
Известно, что водород вызывает охрупчивание некоторых марок стали, но этот газ обычно более совместим с пластиковыми трубами.Тем не менее, остаются опасения по поводу того, может ли водород, который легче метана, вытекать из пластиковых магистралей, предназначенных для транспортировки природного газа. Согласно обзору Market Intelligence, с третьего квартала 2020 года дистрибьюторы газа объявили как минимум о 26 пилотных проектах по водороду, многие из которых сосредоточены на смешивании в трубопроводах.